Haema 2021; 12(1): 5-23
Konstantinos Liapis
Consultant Haematologist, Georgios Gennimatas Hospital
Full PDF | ![]()
⇒See – Cases illustrating the microscopical features of haemoglobinopathies in in: Illustrations of Haemoglobinopathies (Figures 15-21)
⇒See – Haemoglobin electrophoresis and HPLC in Supplementary Appendix 1
The haemoglobinopathies are a common cause of anaemia in the tropics. The most important abnormal haemoglobins in the tropics are Hb S, C, E. The abnormal haemoglobins also occur in Central and South America and the Caribbean, and among blacks in the United States. Hb D has a limited distribution and is clinically mild. Thalassaemia which is a failure of haemoglobin synthesis is also widespread (Figure 1).

Figure 1. Distribution map of haemoglobinopathies and β-thalassaemia.
1.1 THALASSAEMIA
α and β thalassaemia are common in the tropics. The β-thalassaemias are the most important types of thalassaemia because they are so common and more often produce severe anaemia. The frequency of β-thalassaemia (β+, β0) is high in the ‘thalassaemia belt’. The carrier frequency for various forms of β-thalassaemia ranges between 2-30% in this belt (mean 3%). The disease is particularly common in the Mediterranean basin and in South-East Asia, especially, Thailand, Malaysia, and South China.
Although α-thalassaemias are commoner than β-thalassaemias, they pose less of a public health problem because they are less severe. α-thalassaemia is common in sub-Saharan Africa, Saudi Arabia (where, although α+, Ηb Η disease is common due to the presence of non-deletional αΝD or αΤ mutations e.g. αΤsaudi), and particularly common in South-East Asia, parts of India and Nepal, and in the coastal areas of Papua New Guinea where its frequency reaches 70% (Figure 2). In Sri Lanka, 15.5% of the population are α+ and ~3% are heterozygous for ααα (αα/ααα).

Figure 2. Distribution map of α-thalassaemia.
The frequency of α0-thalassaemia genes is highest in South-East Asia and South China e.g. in Thailand, the frequency is 2.5% for α0 (deletional), 10-15% for α+, and 5-15% for Hb Constant Spring (αcsα/). Therefore, Hb H disease constitutes a major health problem in South-East Asia.
α+-thalassaemia gene frequencies are 10-27% in sub-Saharan Africa, but α0 thalassaemia is not present in these populations and, thus, there is no disease (Ηb Barts hydrops fetalis, Ηb Η).
α+-thalassaemia à occurs throughout the tropics and subtropics
α0-thalassaemia (responsible for hydrops fetalis and Ηb Η) à occurs mainly in South-East Asia and, to a lesser extent, in the Mediterranean
Deletional forms of αo-thalassaemia genes:
| – SEA (deletion: 660 bp) | → the ‘South-East Asian’ form of α0-thalassaemia |
| – FIL: (deletion: 550 bp) | |
| – THAI: (deletion: 411 bp) |
– ΜΕD Ι, ΜΕD ΙΙ → the ‘Mediterranean’ form of α0-thalassaemia
Table 1 shows different forms of α-thalassaemia.
Terminology of α-thalassaemia:
αΤ or αND → non-deletional α-thalassaemia
genes e.g. αΤsaudi
-α/ → α+ or α-thalassaemia 2
–/ → α0 or α-thalassaemia 1
Microscopically, α-thalassaemia trait is similar to β-thalassaemia trait except that target cells and basophilic stippling are less prominent. MCH is significantly reduced in both α and β thalassaemia (<27 pg), but MCHC is often normal or nomal/low. Typical values from an individual with α-thalassaemia trait: Ηb 15 g/dl, RBC 5.90×1012/l, Htc 46%, MCV 78 fl, MCH 25.4 pg, reticulocytes 1.2%, WBC 5.2×109/l, PLT 251×109/l.
Hb Η disease is defined as presence of Hb Η on haemoglobin electrophoresis regardless of its percentage. Ηb H disease is characterised by moderate anaemia (Ηb 6-10 g/dl) and splenomegaly. The blood film of a patient with Hb H disease shows marked hypochromia, microcytosis, anisocytosis, marked poikilocytosis, many target cells, hypochromic red cell fragments, leptocytes (severely hypochromic and large cells 10-11 μm with thin border also known as ‘pancake cells’), and dacryocytes, pear-shaped rbcs or tailed rbcs (Figure 3) which disappear after splenectomy. There is also basophilic stippling and polychromasia. The reticulocyte count is increased (5-10%). MCHC is characteristically low. In blood films stained supravitally with brilliant cresyl blue or new methylene blue (i.e. reticulocyte preparations), some of the non-reticulocyte cells are filled with multiple fine, deeply staining deposits which are Hb H inclusions (‘golf-ball’ cells). This finding is specific for Hb H disease.
Note: a falsely elevated platelet count may occur in Hb H due to the presence of marked microcytosis e.g. values from an 28-year-old woman with Hb H: Ηb 8.7 g/dl, Hct 28%, MCV 56.5 fl, MCH 17.2 pg, reticulocytes 9%, MCHC 30.4, RDW 30.3, WBC 6.92×109/l, PLT 1393×109/l (false value; true value was ~300×109/l).
Hb H disease usually results from the inheritance of doubly heterozygous α0-thalassaemia/α+-thalassaemia (i.e. − −/−α), or from the inheritance of α0-thalassaemia and Hb Constant Spring (− −/αcsα), or from the homozygous state for severe non-deletion form of α-thalassaemia particularly common in Saudi Arabia. The forms of Hb Η disease caused by non-deletional mutations (point mutations) are more severe than the classical deletional forms.

Figure 3. Dacryocyte (tear-drop rbc) versus tailed rbc. The dacryocyte (left) has a shorter projection than that of the tailed rbc (right). The tailed rbc is a rbc with an elongated projection with rounded end and constant diametre maintained from beginning to end.
1.2 HAEMOGLOBINOPATHIES
Haemoglobinopathy results from a single mutation of the β gene located in chromosome 11 (β-globin variants):
Hb S: Glut → Val (β6)
Hb C: Glut → Lys (β6)
Hb E: Glut → Lys (β26)
Hb D: Glut → Gly (β121)
Hb O: Glut → Lys (β121)
1.2.1 HAEMOGLOBIN S
The frequency of βS gene in tropical Africa is very high e.g. 15% in Nigeria. This means that a large portion of the population are carriers of sickle cell anaemia (Hb AS) and sickle cell anaemia (Hb SS) is very common. Hb SS is also common in the Caribbean and South America and among blacks in USA (African-Americans) in whom βS gene frequency is 8-10% (Figure 4). There is growing recognition that sickle cell disease represents an increasing global health burden. Although precise estimates of the global number of patients are lacking, the vast majority of this burden occurs in sub-Saharan Africa. The total population of ~100 000 with sickle cell disease in the United States is very small in comparison with the estimated 260 000 children who are born with sickle cell anaemia across sub-Saharan Africa every year.

Figure 4. Haemoglobin S distribution map.
Interaction (co-inheritance) of Hb S with other abnormal haemoglobins may produce mild to severe sickling disorders (Table 2). Of these, Hb SC is particularly common in West and Central Africa (Burkina Faso, Ghana, Benin, northwestern Nigeria). Hb S β+-thalassaemia occurs in West Africa (e.g. Liberia) and in the Mediterranean littoral. Hb S β0-thalassaemia occurs in North Africa, the Mediterranean, and the Caribbean (in the Caribbean, sickling haemoglobinopathies include: Ηb SS 65%, Hb SC 24%, Hb S-β thalassaemia 10%, and other 2%). Hb S β-thalassaemia is characterised by marked hypochromia and numerous target cells. Hb SD occurs in the Punjab (India-Pakistan border).
Sickle cell disease = sickle cell anaemia + other sickling disorders.
With the exception of Hb S β0-thalassaemia, the compound heterozygous genotypes of sickle cell disease are usually less clinically severe than sickle cell anaemia.
Sickle cell anaemia
The typical patient with sickle cell anaemia is seen with a normochromic, normocytic anaemia in which the haemoglobin and red blood cell count are reduced to about one-half normal values.
Note: the number and character of sickle cells on peripheral blood films in patients with sickle cell anaemia may not be sufficiently striking to warrant diagnosis. However, in cases of sickle cell crisis the crescent forms are plentiful. Sickle cells are rarely, if ever, seen in blood films from persons with sickle cell trait (Ηb AS). Sickle cell trait is typically asymptomatic. However, it may be associated with haematuria and, rarely, splenic infarcts at high altitude or after intense exercise.
Sickle cell anaemia is a haemolytic anaemia. The haemolytic nature of anaemia is evidenced on the blood film by the finding of the poikilocytes of haemolysis (spherocytes, red cell fragments, echinocytes, and acanthocytes), and the signs of active red cell regeneration (Howell-Jolly bodies[1], nucleated rbcs, and polychromasia). Reticulocytosis is always present (reticulocytosis may exceed 20%). Target cells are also quite prominent in sickle cell anaemia (++ or +++). PLTs are usually normal but counts >1000×109/l sometimes occur. The leukocyte count is usually normal or increased between 10-16×109/l, but it may be falsely very high during a sickle cell crisis, if there is a large number of circulating nucleated rbcs. In such cases (when 6 or more nucleated rbcs are seen per 100 WBC in the blood film), a correction should be made according to the formula:
Corrected WΒC count (/μl) = WΒC: (100+nRBCs) ×100.
Remember: the presence of many circulating erythroblasts indicates tissue hypoxia!
The classic clinical picture of sickle cell anaemia in tropical Africa is anaemia plus chronic arthralgia or anaemia plus paroxysmal extremity pain. The patients generally have haematocrits 20-25% but feel well with these low values. An important complication of sickle cell anaemia, which is very common in tropical Africa, is the cutaneous ulcer. In fact, some patients >10 years old with sickle cell anaemia may be diagnosed with chronic leg ulceration as their presenting manifestation!
Hb SS and Ηb SC à should be included in the differential diagnosis of tropical ulcer.
Note: there are 2 forms of sickle cells (drepanocytes) as shown in Figure 5:
The crescent form or filamentous form. Characteristics: irreversible sickle cells upon oxygenation (due to polymerisation and precipitation of Hb S); half-moon or crescent shape to cell with centre cell diametre narrow (i.e. dense); more distinct points at each end of cell. This is the typical elongated, narrow-cell-diametre sickle cell.
the oat cell form or boat-shaped cell form. Characteristics: reversible sickle cells upon oxygenation; straight appearance to cell with centre cell diametre broader than filamentous form (i.e. less dense); less distinct points at each end of cell.
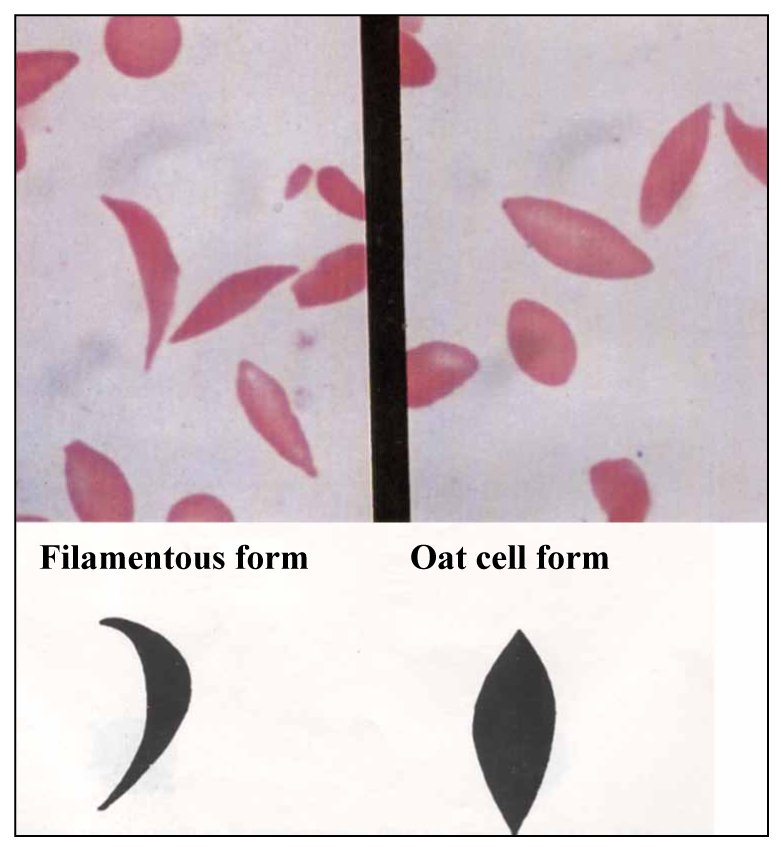
Figure 5. Two forms of sickle cells.
Oat sickle cell forms may cause problems in interpreting blood films, because they may be confused with the elliptocytes of hereditary elliptocytosis, the pencil cells or cigar cells of iron deficiency anaemia, and the envelope cells of Hb C, all of which are common in Africans.
The elliptocyte is an elongated rbc with rounded ends and its cellular diametre appears equivocal from top to bottom. In contrast, the drepanocyte is an elongated rbc with pointed ends and its cellular diametre is broader in the centre of cell and narrows toward the ends of the cell into a point (Figure 6). The pencil cell is an elongated rbc with rounded ends in which its cellular diametre is broader in the centre of cell and narrows toward one end of the cell.
1.2.2 HAEMOGLOBIN C
Hb C occurs almost exclusively in blacks. Hb C is very common in West Africa e.g. Hb C frequency in northern Ghana is 17-28%. It is also common among blacks in the Caribbean and United States (2-3% of African-Americans are A-C heterozygotes).

Figure 6. Elliptocyte versus sickle cell.
A-C heterozygotes (Hb C trait or Hb ΑC) have an Hb A to Hb C ratio of 60%:40%. The blood film shows moderate to high numbers of target cells. They are clinically healthy and have normal rbc indices. They can be diagnosed by chance if their blood film is examined because they have e.g. 30% or 40% target cells!
C-C homozygotes (Hb C disease or Ηb CC) have normal or low MCV and sometimes high MCHC (i.e. dense erythrocytes). Ηb CC is a mild or moderately severe haemolytic anaemia. The patients have splenomegaly, mild or moderately severe anaemia (Hb 9-12 g/dl), and reticulocytes about 4%. In more severe cases, anaemia in Hb CC may be macrocytic due to more pronounced reticulocytosis. Hb electrophoresis at alkaline pH shows a single compenent, slowly migrating to the same position as Hb Α2.
The blood film of patients with haemoglobin C-C disease is characteristic: there are numerous target cells (50-90%) microspherocytes (close inspection shows that the majority are, in fact, irregularly contracted cells), rare crystalline structures that appear as ‘blocks’ or ‘bars of gold’ inside red cells which appear to be otherwise empty of haemoglobin (tetragonal or rectangular crystals of Hb C), and slight to moderate numbers of envelope forms of which there are two types (Figure 7):
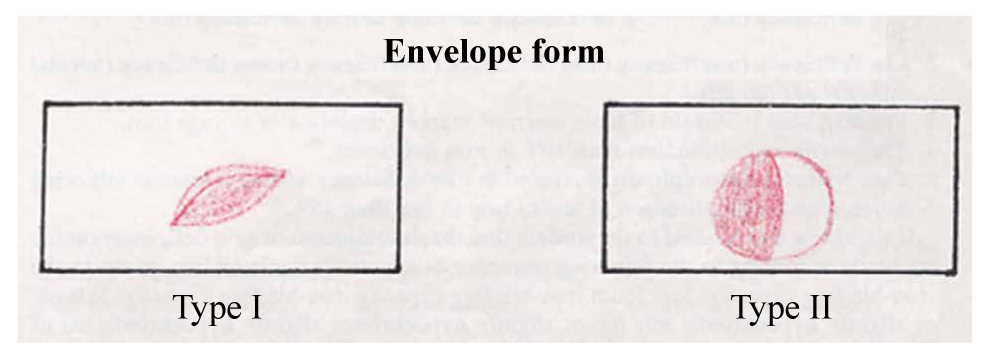
Figure 7. The two types of envelope cells.
Type I which appears to be an erythrocyte folded over upon itself mimicking an envelope and also referred to as a ‘folded cell’ or ‘clam shell’ or ‘clutch pocketbook’ form. It is not seen exclusively in Hb C disease. It is also observed in Hb S disorders.
Type ΙΙ which shows a concentration of haemoglobin to one side in the erythrocyte giving a flap-like appearance to the cell. Type II is also seen in sickling disorders and the thalassaemia syndromes.
Although these envelope forms are not exclusive of Hb C and are seen in other conditions, their number is important. They are usually seen in moderate numbers in Hb C disease, persisting from field to field whereas in other conditions their number is more occasional. The blood film may also show polychromasia and some nucleated rbcs. It is not infrequent for patients with haemoglobin C homozygosity to have microcytosis, often as a result of co-existing α-thalassaemia trait.
Tips:
- numerous target cells and easily seen microspherocytes and type ΙΙ envelope cells within a normochromic, normocytic rbc population à suspect Ηb CC. If significant hypochromia and microcytosis are present à suspect compound heterozygocity of Hb C with β-thalassaemia or with α-thalassaemia (all common in West Africa). Hb electrophoresis will not differentiate Hb CC from Hb C β-thalassaemia because Hb C and A2 migrate together (HPLC is needed).
- if you see a rectangular haemoglobin crystal this is consistent with Ηb CC. Crystals of haemoglobin C are uncommon but when present they indicate Hb CC or Hb C β0-thalassaemia.
Hb C crystallises when its concentration is greater than 44%. Therefore, crystals are not found in Hb C trait (Hb AC). Hb C crystals obstruct the microvasculature but melt in the splenic microenvironment (Hb CC patients may rarely suffer from painful vaso-occlusive events!).
Differential diagnosis of envelope cells:
- stomatocyte: it does not contain concentrated (dense) haemoglobin
- drepanocyte (filamentous form): it is elongated (Figure 8).

Figure 8. Sickle cell versus envelope cell.
Sickle cell/haemoglobin C disease (Ηb SC) is common in West Africa and and is also present among blacks in the Caribbean and USA. Affected individuals have a moderate anaemia (average haemoglobin of 8-10 g/dl), with a slight reticulocytosis, splenomegaly, and a sickling disorder of variable severity. Although the disease is usually less severe than sickle cell anaemia, an individual may experience a painful crisis. The blood film is characteristic: high numbers of target cells, few reversible sickle cells (oat cells), and envelope cells (folded cells), and some bizarre cells with a peculiar crystal shaped like ‘a gloved hand’ or a pointed crystal like the ‘Washington Monument’ (SC poikilocytes). Hb electrophoresis shows 50% Hb S and 50% Hb C.
Hb C may combine with β-thalassaemia to produce haemoglobin C/β-thalassaemia, which may be symptomatic because of anaemia. Τhe clinical picture is dependent on whether the thalassaemia is β+ or β0 type. Ηb C β+-thalassaemia is commoner in West African populations (e.g. Liberia) and the clinical disorder is usually mild; features include low-grade anaemia, low MCV, minimally elevated reticulocytes, Ηb Α2 >3.5%, and absent or some splenomegaly. Ηb C β0-thalassaemia leads to a moderately severe anaemia with splenomegaly and/or hypersplenism (the clinical picture may resemble thalassaemia intermedia). The blood film shows numerous target cells, microspherocytes (or irregularly contracted cells), marked microcytosis, and hypochromia. There is more anisocytosis, microcytosis, and poikilocytosis than in haemoglobin C disease, particularly in Hb C/β0-thalassaemia. Haemoglobin C crystals are sometimes present (mainly in Ηb C β0-thalassaemia).
Note: the blood film in haemoglobin C/β-thalassaemia may be indistinguishable from that in haemoglobin C disease. and shows microspherocytes and target cells. A very low MCV value is a clue to diagnosis. Hb C/β-thalassemia is common in African populations.
1.2.3 HAEMOGLOBIN Ε
Hb E is extremely common in South-East Asia (its frequency is highest in Cambodia, Laos and Thailand where it may reach 50-60%, and lower in Indonesia, the Philippines, Malaysia, Singapore, and Vietnam); it also occurs in India, Sri Lanka, and Pakistan. A-E heterozygotes (Hb E trait or Ηb ΑΕ) are asymptomatic (the haemoglobin distribution on Hb electrophoresis is 70-80% Ηb A and 20-30% Ηb E). The blood count shows a minor reduction of MCV and MCH. The blood film may be normal or show microcytosis or few target cells or irregularly contracted cells. E-E homozygotes (Hb E disease or Ηb ΕΕ) may have a mild haemolytic anaemia and splenomegaly, or be asymptomatic. Blood films show a marked microcytic hypochromic picture, numerous target cells (characteristic finding), slight polychromasia, and occasional irregularly contracted erythrocytes (not as many as in Hb CC). Irregularly contracted cells occur because Hb E is an unstable haemoglobin capable of precipitation. Basophilic stippling may be present (not as prominent as in β-thalassaemia). Overall, the microcopical picture in is very similar to β-thalassaemia trait! The blood count is also quite similar to β-thalassaemia trait with a mild anaemia or a normal Hb, elevated RBC number, and reduced MCV and MCH. The reticulocyte count is usually normal.
There are no specific morphological findings in Hb E as in Ηb Η, Ηb S, and Ηb C. On alkaline electrophoresis, there is a strong band (90%) located in the same position as Hb A2 (Hb E 90%, Hb F 5-10%).
Hb Ε is very important because when combined with β-thalassaemia i.e. co-inheritance with β-thalassaemia trait (Hb E/β-thalassaemia compound heterozygocity; genotype βΕ/βthal, either βΕ/β+ or βΕ/β0) may lead to the clinical picture of thalassaemia intermedia or transfusion-dependent anaemia (βΕ/β+ produces a milder anaemia). Hb E β-thalassaemia is a serious problem in South-East Asia and India.
Co-inheritance of Hb E and α-thalassaemia trait (either α1 or α2 heterozygotes) does not cause problem (these cases have less Hb E e.g. 17% than 30%). Individuals with co-inheritance of Hb E and Hb H (either α1/α2 or Constant Spring disease i.e. –/αcsα) have symptomatic anaemia and splenomegaly.
Remember:

1.3 G6PD DEFICIENCY
There are several variants of G6PD deficiency depending on residual G6PD enzymatic activity. The most common variants in the tropics are:
| G6PD variant: | G6PD activity: |
| Mediterranean (GdMediterranean) | <10% of normal activity |
| Union (GdUnion) | <10% of normal activity |
| Α- (Αfrican or GdA-) | 10-60% of normal activity |
| Canton (GdCanton) | <10% of normal activity |
| Mahidol (GdMahidol) | 10-60% of normal activity |
There are three geographical zones of G6PD deficiency (Figure 9):

Figure 9. Percentage (%) of male population that is G6PD deficient (according to WHO).
Ζone I (Mediterranean, Middle East, Arabian Peninsula, India): Mediterranean variant. Clinical manifestations: favism; drug-induced haemolysis (e.g. antimalarials, dapsone, sulfonamides, quinolones); infection-induced haemolysis (e.g. hepatitis, typhoid, malaria).
Ζone ΙΙ (South-East Asia): Μediterranean, Canton, Union, Mahidol variants. Clinical manifestations: favism; drug-induced haemolysis (e.g. antimalarials, dapsone, sulfonamides, quinolones).
Ζone III (tropical Africa in blacks): Α- variant which may cause mild drug-induced haemolysis e.g. antimalarials, dapsone. In the haemolysis of GdA- variant, it is sometimes possible to continue with essential therapy, for example dapsone for treatment of leprosy or primaquine for P. vivax. These patients have a transient haemolytic anaemia, followed by a state of compensated haemolysis.
Two variants (G6PD A- and G6PD Mediterranean) are the most common worldwide. G6PD A- has an occurrence of 10% in blacks (Africans and African-Americans). G6PD Mediterranean is also prevalent in the Middle East. Favism is unknown in tropical Africa and South America.
The morphological findings of G6PD deficiency–related acute haemolytic anaemia include the poikilocytes of haemolysis (spherocytes, red cell fragments, echinocytes, and acanthocytes), the signs of active red cell regeneration (Howell-Jolly bodies, nucleated rbcs, fine basophilic stippling, polychromasia), and the specific signs of oxidative haemolysis (blister cells, bite cells, irregularly contracted cells, and Heinz bodies). Blister cells are also called hemighosts or eccentrocytes. In addition, ghost cells or phantom cells (i.e. erythrocytes completely devoid of haemoglobin) may be seen in severe G6PD deficiency-related haemolysis. The Heinz bodies (large aggregates of denatured haemoglobin) are visible on methyl violet or crystal violet supravital staining (Figure 10).
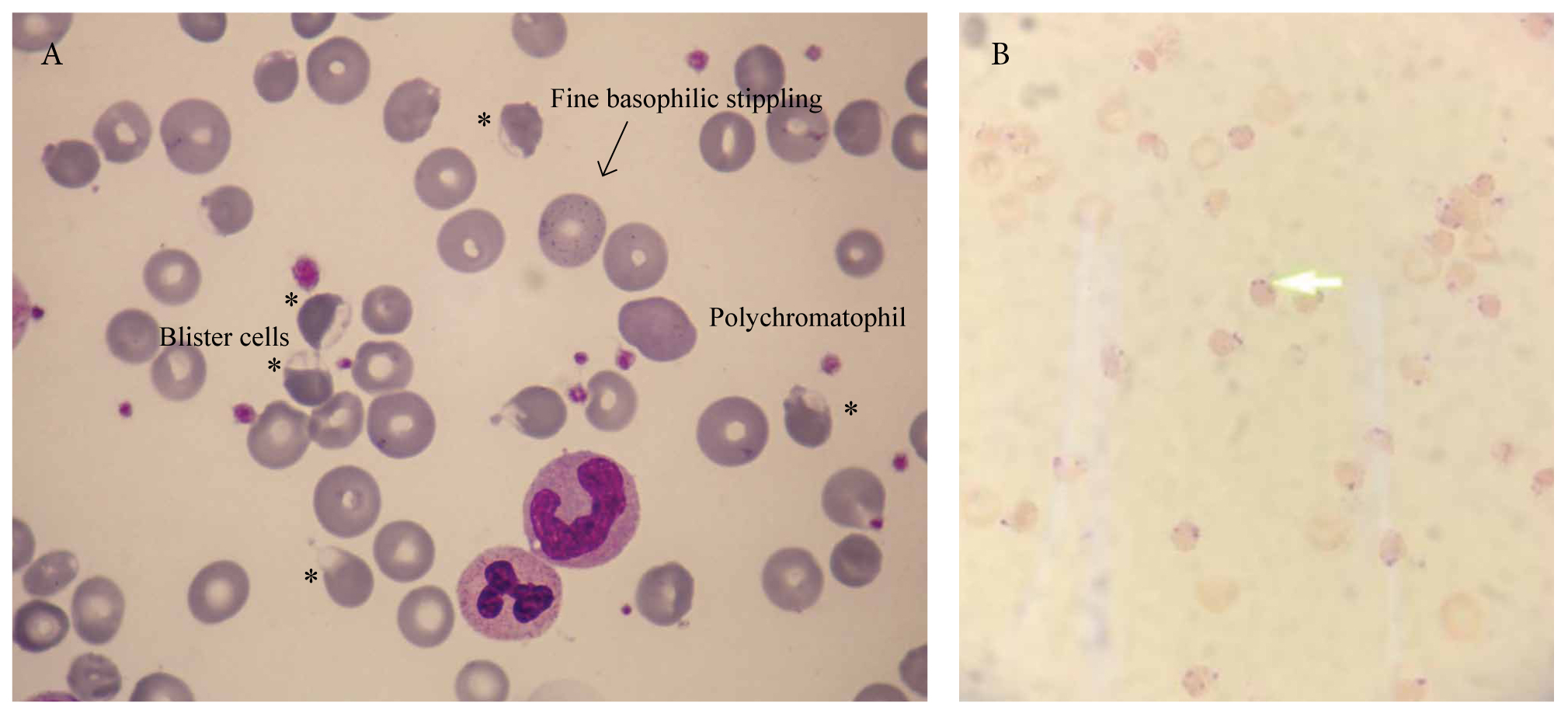
Figure 10. Favism in a 66-year-old Greek man who ate fava beans one and two days earlier. Panel A shows the characteristic morphological findings of favism. Panel B shows Heinz bodies (methyl violet supravital stain).
The differential diagnosis of oxidative haemolysis (i.e. irregularly contracted cells and/or blister cells; detectable Heinz bodies) includes:
- G6PD deficiency
- Ηb CC
- unstable haemoglobins e.g. Hb Bristol, Hb Köln, Hb Zürich
- Wilson’s disease
- Zieve’s syndrome
- oxidant-induced haemolysis: significant exposure to oxidant drugs or chemicals (intoxication or chronic use)
Figure 11 illustrates an example of oxidative haemolysis.

Figure 11. A and B. Oxidative haemolysis: irregularly contracted cells (arrows), microspherocytes (+), blister cells (*), polychromatophils (>>).
Tips:
- large numbers of blister cells are characteristic of the Mediterranean variant whereas in blacks the typical findings are irregularly contracted cells and bite cells with few blister cells.
- individuals with haemolytic anaemia due to G6PD deficiency may also develop methaemoglobinaemia.
- small or sometimes moderate numbers of blister cells may be seen in sickle cell anaemia, especially in cases complicated by pulmonary infarction. Co-inheritance of G6PD deficiency should be ruled out.
1.4 SOUTH-EAST ASIAN OVALOCYTOSIS AND AFRICAN ELLIPTOCYTOSIS
In the areas shown in Figure 12, the prevalence of hereditary ovalocytosis and hereditary elliptocytosis is high (>2%).

Figure 12. Distribution of South-East Asian ovalocytosis and elliptosytosis.
South-east Asian ovalocytosis (SAO): The molecular basis is a deletion of 27 nucleotides, resulting in the deletion of nine amino acids from band 3. The abnormal SAO band 3 has a higher than normal affinity for ankyrin, resulting in increased membrane rigidity and the oval shape. Inheritance is autosomal dominant. All affected individuals are heterozygous (homozygosity is probably lethal). Ovalocytosis is seen at high frequency (up to 27%) in Melanesian populations (e.g. Papua New Guinea, Solomon Islands, Torres Strait Islands) and to lesser extent in the Philippines, Malaysia, and Indonesia.
SAO does not usually cause haemolysis or anaemia. Individuals are usually completely asymptomatic unless there is an additional reason for anaemia e.g. iron deficiency, megaloblastic anaemia, or intercurrent malaria (these complicated cases may cause problems in the interpretation of the blood film).
The blood film is pathognomonic: a high proportion of red cells are ovalocytes (elongated cells with a long axis less than twice the transverse axis ≠ elliptocytes), stomatocytes and knizocytes (with duplicated central pallor like Ɵ or rarely triplicated central pallor like O), several macro-ovalocytes (with almost twice the size of other cells), and stomatocytic cells with longitudinal or transverse stomas, V-shaped stomas, Y-shaped stomas (‘cat mouth’ like), or slits of pallor instead of the typical central linear mouth-like slits (Figure 13). Knizocytes Ɵ may also be observed in thalassaemia, liver disease, megaloblastic anaemia, myelodysplasia, and congenital dyserythropoietic anaemia. Knizocytes O, although not specific, are more characteristic of SAO. Osmotic fragility is reduced (i.e. rigid membrane). The differential diagnosis includes hereditary elliptocytosis, hereditary stomatocytosis and Rh null disease, liver disease, alcoholism, and megaloblastic anaemia.
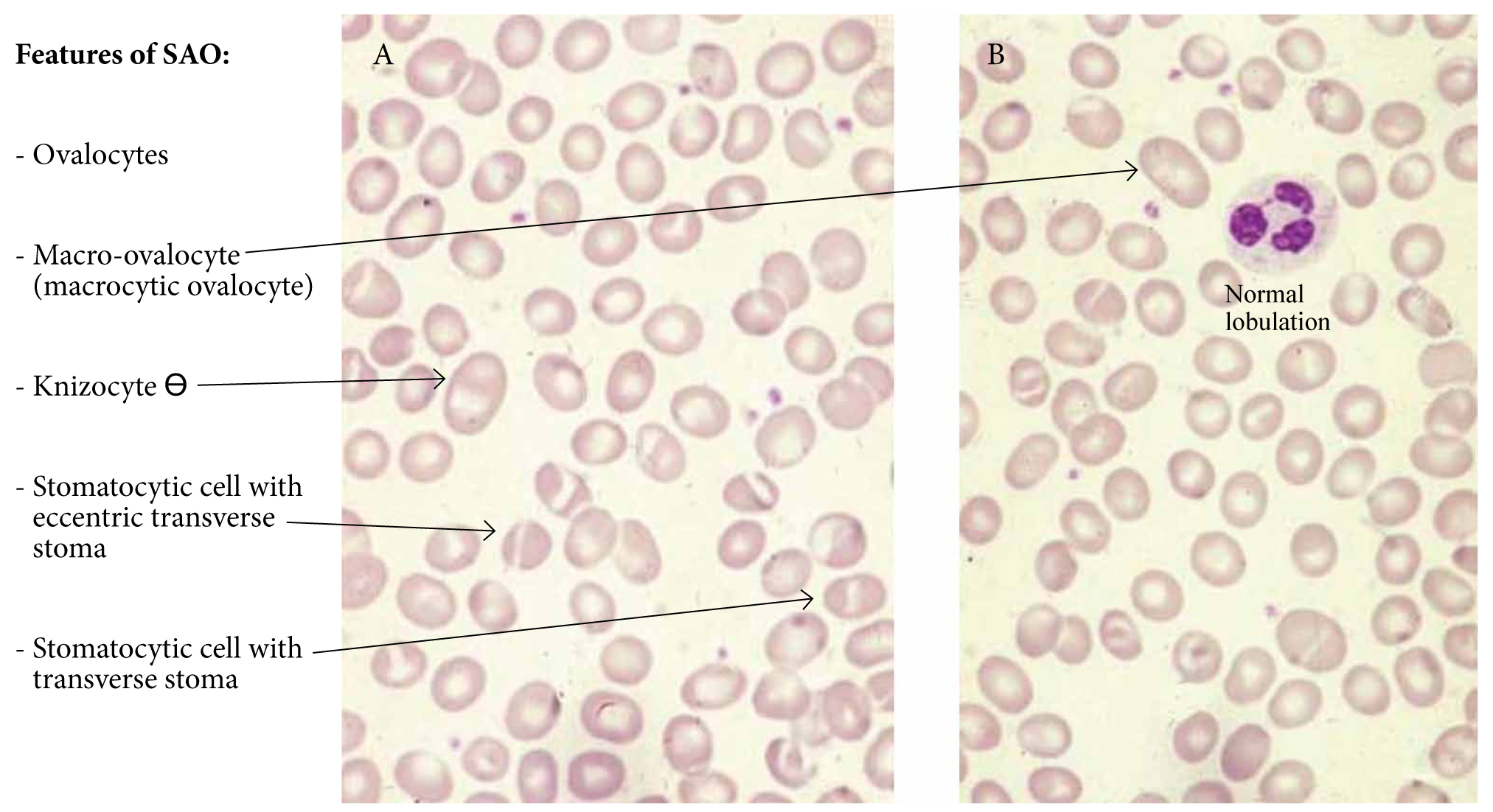
Figure 13. Blood film of South-East Asian ovalocytosis.
Αfrican elliptocytosis: Hereditary elliptocytosis is more common in West and North Africa. Up to 2-3% of both northern and southern Nigerians have an uncharacterised elliptocytosis. It is caused by mutations in spectrin. Inheritance is autosomal dominant. The condition is usually symptomless, but there may be mild haemolysis. The homozygous condition which is called hereditary pyropoikilocytosis, is a form of chronic haemolytic anaemia with splenomegaly.
The blood film in hereditary elliptocytosis is pathognomonic: it shows numerous elliptocytes (++++). The blood film in hereditary pyropoikilocytosis shows an extreme picture: microspherocytes ++++, poikilocytes ++++, fragmented rbcs, many ovalocytes and elliptocytes, nucleated rbcs, and polychromasia (Figure 14).

Figure 14. Panel A shows African elliptocytosis. Panel B shows hereditary pyropoikilocytosis.
Remember: the elliptocyte is an elongated cells with a long axis more than twice the transverse axis (≠ ovalocyte), with rounded ends (≠ drepanocyte), and its cellular diametre appears equivocal from top to bottom (≠ pencil cell/cigar cell, drepanocyte). For diagnosis of elliptosytosis, >10% elliptosytes are required.
1.5 ILLUSTRATIONS OF HAEMOGLOBINOPATHIES
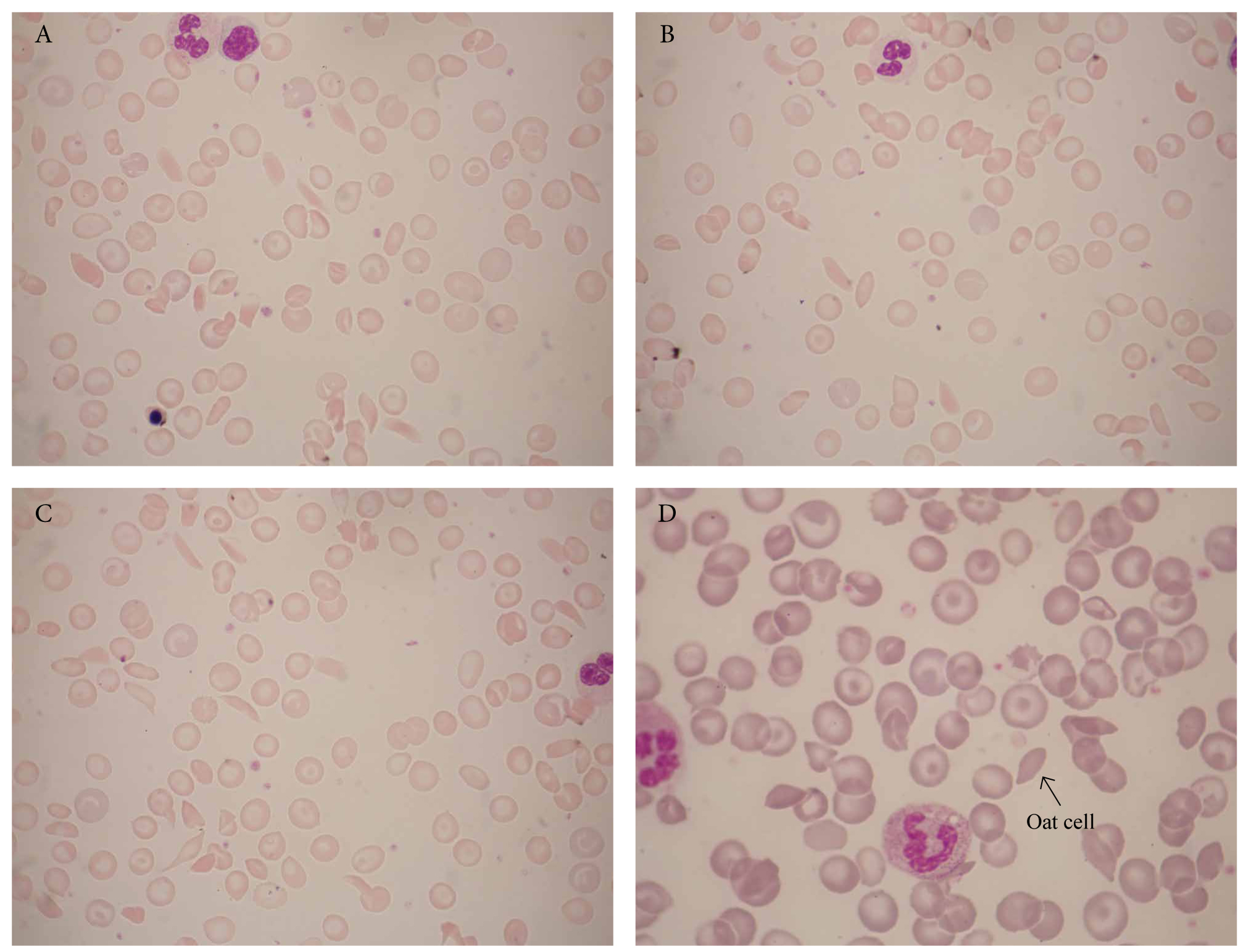
Figure 15. Sickle cell anaemia (Ηb SS). Panels A-C illustrate a typical case of sickle cell anaemia showing crescent forms, oat cells, target cells, envelope cells, polychromasia, and a nucleated rbc. Panel D is a less typical case of sickle cell anaemia. Both patients were from Ghana.
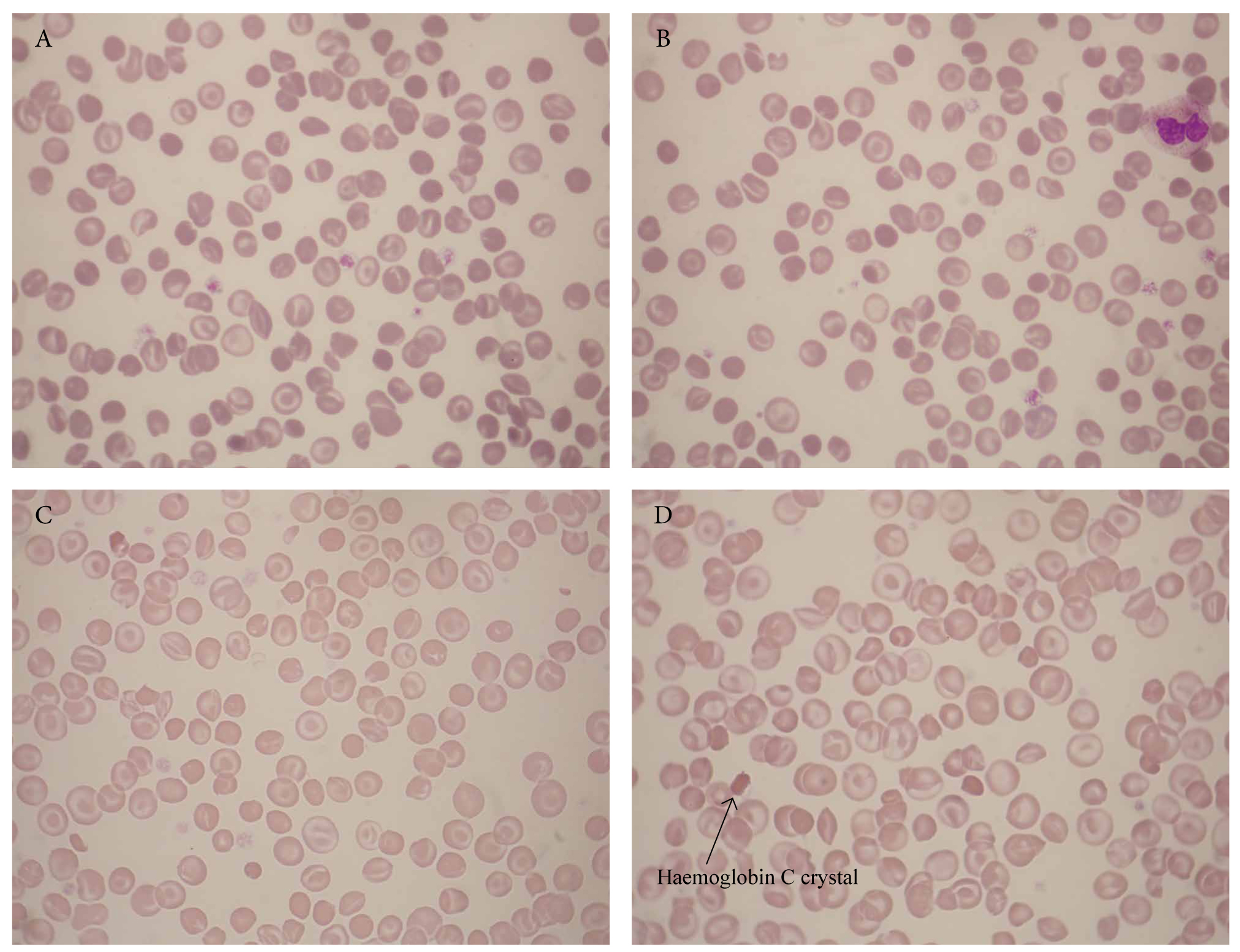
Figure 16. Blood films of two patients with Hb C disease (Ηb CC) showing target cells, irregularly contracted cells, microspherocytes, type I and type II envelope cells, and an Hb C crystal (arrow). The first patient (A and B) was from Nigeria; the second patient (C and D) was from the Caribbean.
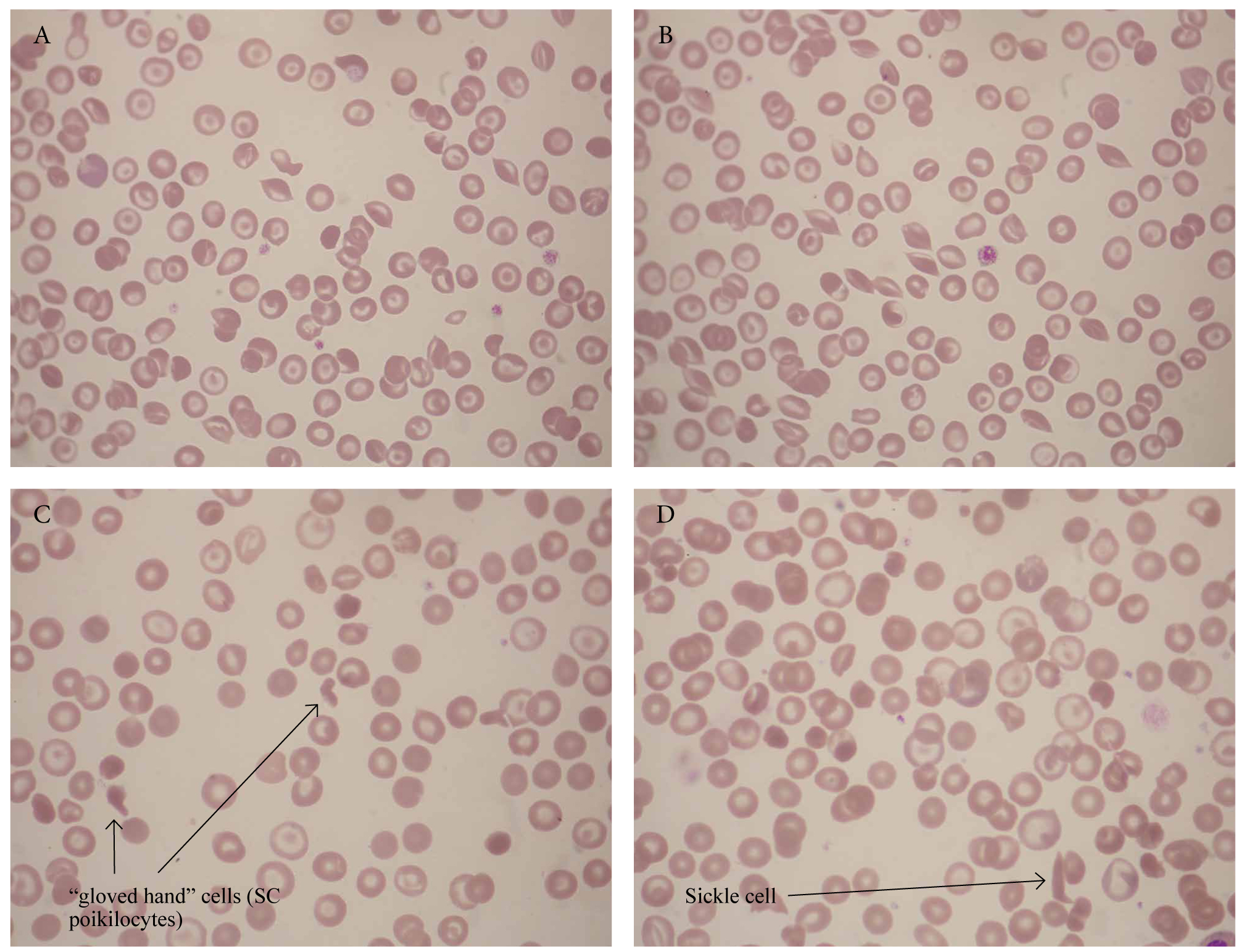
Figure 17. Two cases of S-C disease (Ηb SC) showing target cells, irregularly contracted cells, envelope cells, pointed ends, and SC poikilocytes. Both patients were from the Caribbean: the first patient (A and B) was from the Antilles and the second patient (C and D) from Jamaica.
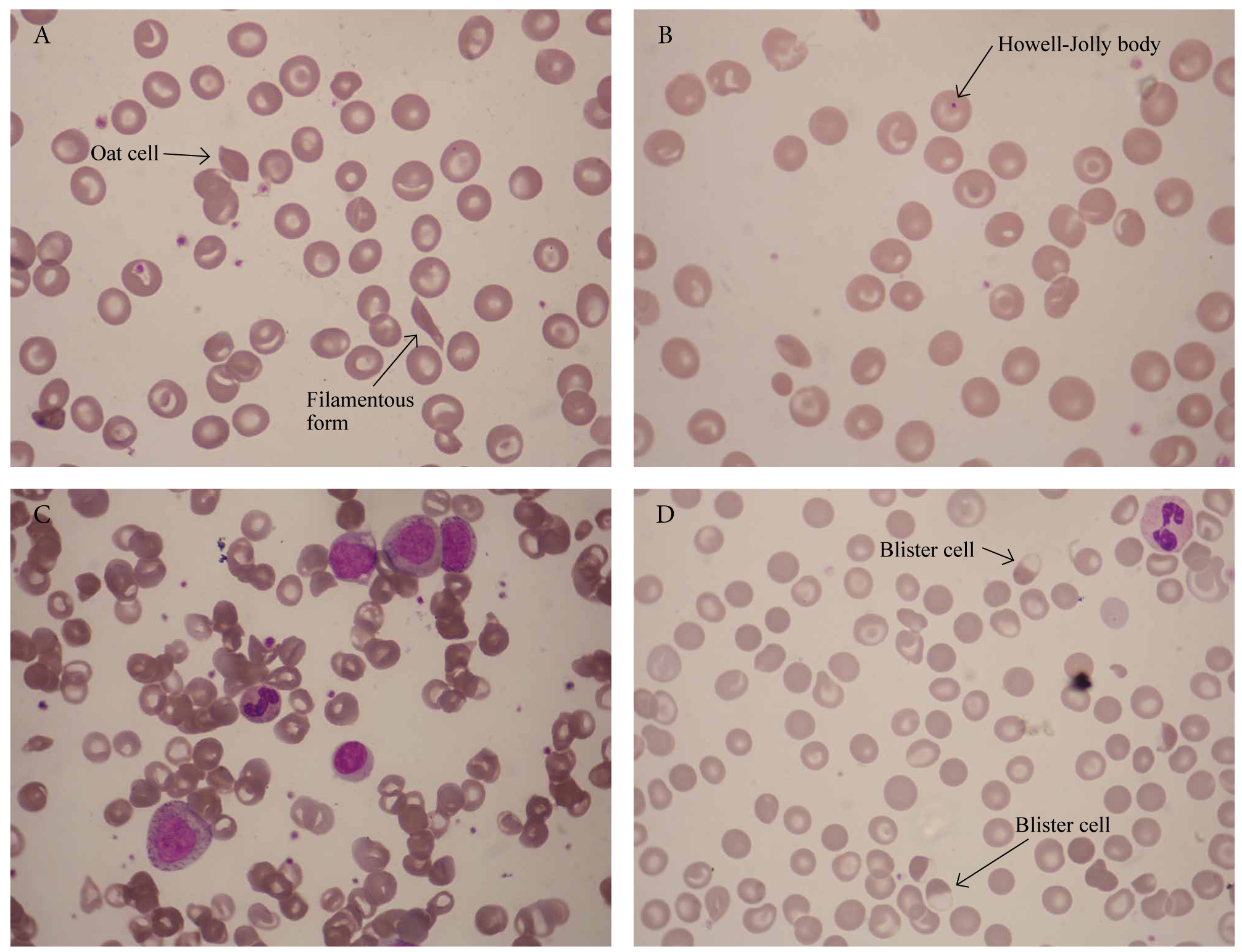
Figure 18. Blood film of a 37-year-old Greek man with sickle cell anaemia and G6PD deficiency, fever, cough, bone pain, and right lower lobe pneumonia. Hb 6.4g/dl, Hct 19.4%), MCV 76 fl, MCH 28.4 pg, ΜCHC 32.9, RDW 15.6, WBC 12.4×109/l, PLT 203×109/l, reticulocytes 4.4%. Treatment started with levofloxacin. Panels A and B illustrate sickle cells (crescent forms and oat cells), target cells, Howell-Jolly bodies, microspherocytes. Panel C shows neutrophil leukocytosis with myelocytes (‘left shift’). Four days after admission, he developed worsening anaemia and jaundice. Blister cells due to G6PD deficiency-related haemolysis induced by levofloxacin (panel D).

Figure 19. Blood film of a 13-year-old boy who presented with severe generalized joint pain of one day’s duration. No fever, chills, vomiting, weight loss. The painful joints were neither swollen, warm, nor tender to palpation. Hb 6.8 g/dl, WBC 13.0 × 109/l, PTL 569 × 109/l. Sickle cell crisis: sickle cells +++, boat cells ++, target cells +, pointed ends of red cells like thin threads. His parents were from Ghana.

Figure 20. Blood film (A-D) of a 6-year-old boy with Hb C trait. Hb 9.9 g/dl, WBC 9.7×109/l, Hct 30%. Target cells +++. His mother was from the Caribbean.

Figure 21. Blood film (A-D) of a 3-month-old girl from India with β-thalassaemia major. Hb 6.6 g/dl, RBC 6.6 × 1012/l, MCV 57 fl, MCH 21 pg, MCHC 20.3, HbA2 5.3%, HbA nil, HbF ++. Microcytosis +++, hypochromia +++, poikilocytosis +++, anisocytosis +++, tear-drop cells ++, target cells, polychromatophils, nucleated rbcs. Family study: mother: Hb 12.9 g/dl, RBC 6.4 × 1012/l, MCV 61.3 fl, MCH 20.1 pg, MCHC 32.8 (β-thalassaemia trait); father: Hb 14.3 g/dl, RBC 7.0 × 1012/l, MCV 61.0 fl, MCH 20.4 pg, MCHC 33.3 (β-thalassaemia trait); brother: Hb 12.7 /dl, RBC 6.7 × 1012/l, MCV 55.9 fl, MCH 18.7 pg, MCHC 33.6 (β-thalassaemia trait).
Faced with a difficult diagnosis or uncertainty, physicians should consult with haematologists who have expertise in the field of microscopical diagnosis of tropical diseases.
I have found the following references of considerable value in preparing this manuscript. Many further references will be found in each of these works.
References
- Peters W, Gilles HM. A colour atlas of tropical medicine and parasitology. 2nd ed. London: Wolfe; c1981.
- Peters W, Pasvol G. A colour atlas of tropical medicine and parasitology. 6th ed. London: Mosby Elsevier; c2007.
- Cook GC, Zumla AI. Manson’s tropical disease. 22nd ed. London: Εlsevier Saunders; c2008.
- Farrar J, Hotez P, Junghanss T, Kang G, Lalloo D, White N. Manson’s tropical diseases. 23rd ed. London: Εlsevier Saunders; c2014.
- Bain BJ. Blood cells – a practical guide. 3rd ed. London: Blackwell Publishing; c2002.
- Kapff C, Jandl J. Blood atlas and sourcebook of hematology. 2nd ed. Boston: Little Brown and Company; c1991.
- Bain BJ. Haemoglobinopathy diagnosis. 2nd ed. London: Blackwell Publishing; c2006.
- O’Connor BH. A color atlas and instruction manual to peripheral blood cell morphology. 1st ed. Philadelphia: Lippincott Williams & Wilkins; c1984.
- Steinberg MH, Forget BG, Higgs DR, Weatherall DG. Disorders of Hemoglobin. 2nd ed. Cambridge: Cambridge University Press; c2009.
- World Health Organization. Βench aids for the morphological diagnosis of anaemia. Geneva: WΗO; c2001.
- Liapis K, Charitaki E, Delimpasi S. Hemolysis in Wilson’s disease. Ann Hematol. 2011 Aug; 90(4):477-8.
- Halabi-Tawil M, Lionnet F, Girot R, et al. Sickle cell leg ulcers: a frequently disabling complication and a marker of severity. Br J Dermatol. 2008 Feb;158(2):339-44.
- Fairhurst RM, Casella JF. Homozygous hemoglobin C disease. N Engl J Med. 2004 Jun;350(26):e24.
- Schrier SL, Bunyaratvej A, Khuhapinant A, Fucharoen S, Aljurf M, Snyder LM, et al. The unusual pathobiology of hemoglobin Constant Spring red blood cells. Blood. 1997 Mar; 89(5):1762-9.
- Luzzatto L, Arese P. Favism and glucose-6-phosphate dehydrogenase deficiency. N Engl J Med. 2018 Jan;378(1):60-71.
- Cappellini MD, Fiorelli G. Glucose-6-phosphate dehydrogenase deficiency. Lancet. 2008 Jan;371(9606):64-74.
- Piel FB, Williams TN. Subphenotypes of sickle cell disease in Africa. Blood. 2017 Nov;130(20):2157-8.
- Piel FB, Steinberg MH, Rees DC. Sickle cell disease. N Engl J Med. 2017 Apr376(16):1561-73.
- Ware RE, de Montalembert M, Tshilolo L, Abboud MR. Sickle cell disease. Lancet. 2017 Jul;390(10091):311-23.
- Kato GJ, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev. 2007 Jan;21(1):37-47.
[1] Howell-Jolly bodies are also a feature of functional hyposplenism in sickle cell anaemia.