Haema 2021; 12(1): 39-41
Konstantinos Liapis
Consultant Haematologist, Georgios Gennimatas Hospital
Full PDF | ![]()
- Haemoglobin electrophoresis in cellulose acetate at alkaline pH (cellogel; pH=8.3 or pH=8.6). At alkaline pH, haemoglobins are negatively charged proteins so they move toward the anode (+), as shown in Figure S1.

Figure S1. Alkaline haemoglobin electrophoresis.
- Haemoglobin electrophoresis in agarose citrate at acid pΗ (agarose gel, pH=6.0 or pH=6.2 or pH=6.5). At acid pH, haemoglobins are positively charged proteins so they migrate toward the cathode (-), as shown in Figure S2.

Figure S2. Acid haemoglobin electrophoresis.
At alkaline pΗ, haemoglobins S, D, G migrate together at the same position. Hb Lepore (δ-β fusion hybrids) migrates very close to S/G/D. Their distinction is possible with electrophoresis at acid pH, HPLC and sickling test. Three Lepore haemoglobins have been identified on the basis of δ-β crossover: Lepore-Boston (also called Lepore-Washington or Hb Pylos), Lepore-Hollandia, and Lepore-Baltimore. Hb Lepore results in a β-thalassaemia-like condition: heterozygous Hb Lepore resembles thalassaemia minor and the homozygous state results in a thalassaemia major-like condition. Hb D has a limited distribution (Punjab region at India-Pakistan border, where its incidence is 3%) and is clinically mild. Hb D heterozygotes are completely asymptomatic; Hb D homozygotes have mild anaemia with many target cells in the blood film or they are asymptomatic. Hb G is a rare α chain variant seen in Ghana and in African-Americans (Ηb GPhiladelphia). Hb G is stable and is not associated with haematological abnormalities.
There are 6 haemoglobins associated with the sickling phenomenon except Hb S (they all have the mutation β6: GlutàVal plus one additional point mutation): Ηb CHarlem, Hb CGeorgetown, Hb SAntilles, Hb SOman, Hb STravis, and Hb SProvidence. They are associated with a (+) sickling test and (+) solubility test, but migrate at a different position on alkaline Hb electrophoresis and HPLC. Clinically, these haemoglobins behave as Hb S.
Hb I (an α chain variant, stable, no symptoms) and a large quantity of Hb Barts (γ4) may give a (+) solubility test. The clinical importance of Hb I is that it migrates at the same position as Hb Η in alkaline electrophoresis (fast Hb variant). Hb I is not associated with Hb H inclusions or golf-ball cells. Hb I is found in the Mediterranean littoral and in Africa.
Hb OArab is rare in the tropics. Hb O is a β haemoglobin variant: Glut à Lys (β121). Hb O is characterised by the formation of denser and more spherical erythrocytes, leading to elevated MCHC in combination with a slight decrease in MCV. The clinical importance of this haemoglobin is that it migrates at the same position as Hb C in alkaline Hb electrophoresis, but they are separated on acid Hb electrophoresis. Haemoglobin O-Arab heterozygotes show no clinical manifestations; homozygotes present with mild haemolysis and splenomegaly of minimal clinical significance, but may develop haemolytic anaemia during infection or severe illness. Importantly, the anaemia caused by combinations of Hb O-Arab with β thalassaemia trait (β+ or β0) varies from benign to transfusion-dependent, and sickling is enhanced when Hb S and Hb OArab coexist. Although Hb OArab is widely distributed, it is mostly detected in Eastern Mediterranean and Middle East populations. The Greek Pomaks, a Muslim population of the mountainous area of Thrace, demonstrate Hb OArab in impressively high percentage (5.076%), which reaches 27.4% in selected villages (Hb OThrace).
- Cation-exchange High Performance Liquid Chromatography (HPLC)
The normal HPLC pattern is shown in Figure S3.
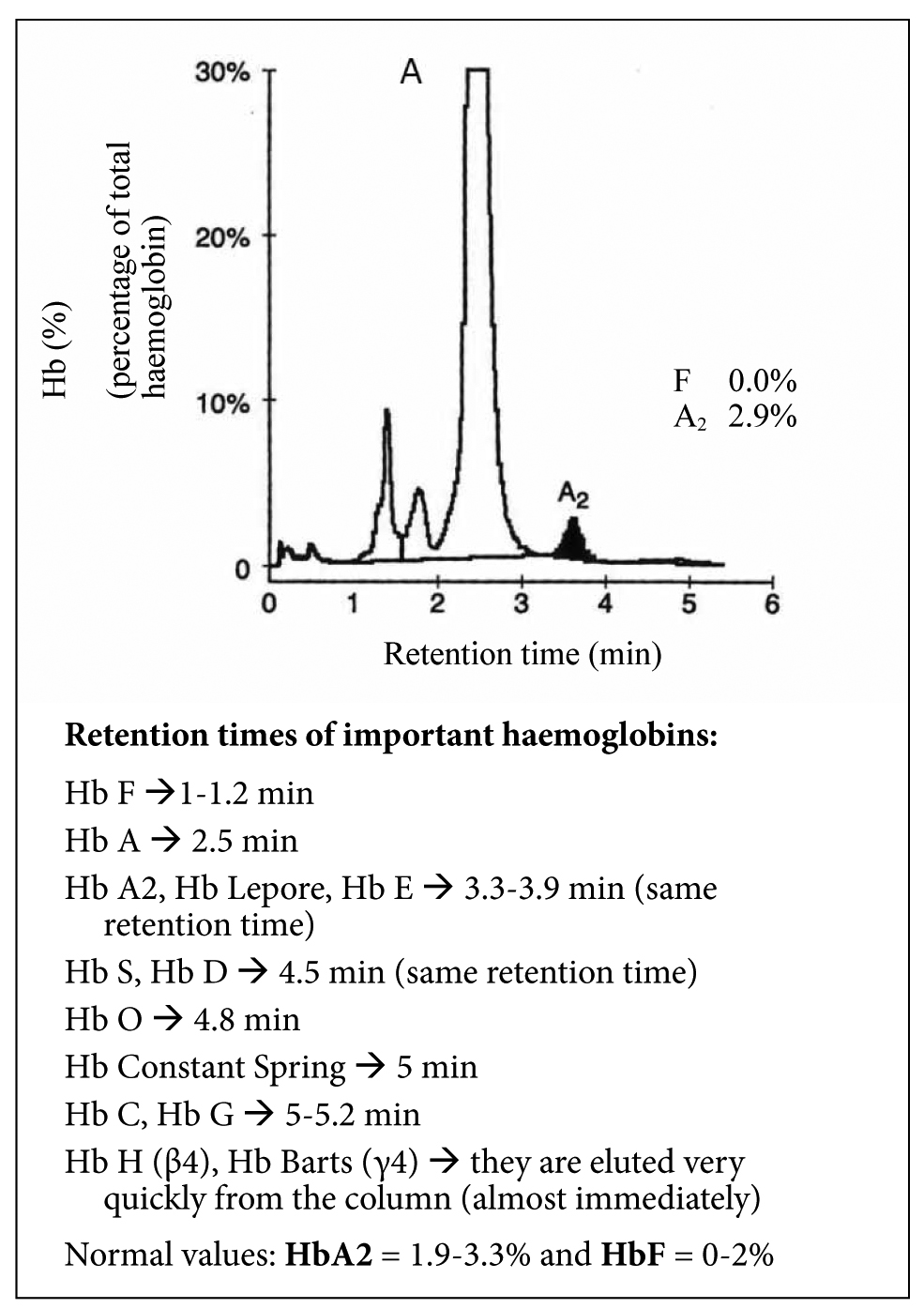
Figure S3. Normal HPLC pattern.
Normal values: HbΑ2 = 1.9-3.3% and HbF = 0-2%
Example 1: A 13-year-old girl of Filipino descent, with hypochromia, microcytosis, and many target cells. No history of transfusion and her parents are healthy. Figure S4 shows her HPLC. Diagnosis: Hb Ε heterozygote (Hb AΕ).
Example 2: A 33-year-old man from Νigeria with anaemia (Hb 10.0 g/dl, MCV 82 fl), splenomegaly and recurrent leg pain. No history of transfusion. His family history is unknown. Figure S5 shows his HPLC. Diagnosis: Hb SC disease.

Figure S4. HPLC consistent with heterozygous HbE (HbAE).
B. S. Haldane first suggested that that the geographical co-incidence of malaria and β-thalassaemia major (Cooley’s anaemia) could be due to the heterozygotes (β-thalassaemia minor) being at genetic advantage through a partial protection against P. falciparum. A relative resistance to malaria was confirmed in Liberian children with thalassaemia minor (β/β+). Another classic example of what Haldane called balanced polymorphism (i.e. heterozygotes are protected against malaria while the harmful genetic effects are restricted to homozygotes) is Hb S. African children who are heterozygous for Hb S are 10 times less likely to develop life-threatening complications of P. falciparum infection than those who lack this allele.

Figure S5. HPLC consistent with HbSC disease.
Tips:
- Always obtain a family history when haemoglobinopathy or thalassaemia is suspected!
- The diagnosis of heterozygous β-thalassaemia (β-thalassaemia minor) depends upon finding an increased Hb A2 >3.5%, usually 4-6% (a higher value may be seen in some cases but values of Hb A2 >7% are rare). – -Hb F is slightly increased in 40-50% of individuals with heterozygous β-thalassaemia (usually up to 3%; in β/β0 trait up to 5%). In cases of:
-HbF >5% → consider δβ-thalassaemia carrier (Hb A2 <3%) or HPFH heterozygote (Hb F 5-16%).
-low Hb Α2 (<1.9%) → consider co-inheritance of δ-thalassaemia
-Hb A2 ≥19% → consider Hb Ε (Hb Ε migrates at the same position as HbΑ2 on alkaline and acid Hb electrophoresis and HPLC). - In carriers of sickle cell anaemia (Hb AS), the percentage of Ηb S is usually 35-45% (because the rate of Ηb S synthesis is slower than Hb Α). If:
-Hb S is <33% → consider S-α thalassaemia co-inheritance.
-Hb S is ≥50% → consider S-β thalassaemia (also has an increased Hb A2 3.5-5% and HbF 5-10% or more) or sickle cell anaemia and recent blood transfusion.
I have found the following references of considerable value in preparing this manuscript. Many further references will be found in each of these works.
References
- Lewis SM, Bain B, Bates I. Dacie, Lewis’s Practical Haematology, 9th ed. Edinburgh: Churchill Livingstone; c2001.
- Bain BJ. Haemoglobinopathy diagnosis. 2nd ed. London: Blackwell Publishing; c2006.
- Steinberg MH, Forget BG, Higgs DR, Weatherall DG. Disorders of Hemoglobin. 2nd ed. Cambridge: Cambridge University Press; c2009.
- Joutovsky A, Hadzi-Nesic J, Nardi MA. HPLC retention time as a diagnostic tool for hemoglobin variants and hemoglobinopathies: a study of 60000 samples in a clinical diagnostic laboratory. Clin Chem. 2004 Oct;50(10):1736-47.
- Khera R, Singh T, Khuana N, Gupta N, Dubey AP. HPLC in characterization of hemoglobin profile in thalassemia syndromes and hemoglobinopathies: a clinicohematological correlation. Indian J Hematol Blood Transfus. 2015 Mar;31(1):110-5.
- Luzzatto L. Haemoglobinopathies including thalassaemia. Part 3. Sickle cell anaemia in tropical Africa. Clin Haematol. 1981 Oct;10(3):757-84.
- Colombo B, Martínez G. Haemoglobinopathies including thalassaemia. Part 2. Tropical America. Clin Haematol. 1981 Oct;10(3):730-56.
- Wasi P. Haemoglobinopathies including thalassaemia. Part 1: Tropical Asia. Clin Haematol. 1981 Oct;10(3):707-29.
- Voskaridou E, Konstantopoulos K, Kollia P, Papadakis M, Loukopoulos D. Hb Lepore (Pylos)/Hb S compounds heterozygosity in two Greek families. Am J Hematol. 1995 Jun; 49: 131-4.
- Papadopoulos V, Dermitzakis E, Konstantinidou D, et al. The origin of Greek Pomaks based on HbO-Arab mutation history. Haema. 2006 Oct; 9(3):380-394.
- Papadopoulos V, Vassiliadou D, Xanthopoulidis G, Petridis D, Agorasti A, Loukopoulos D. The implications of haemoglobin O-Arab mutation. Haema. 2003; 6(4): 296-303.