Haema 2021; 12(1): 30-38
Konstantinos Liapis
Consultant Haematologist, University Hospital of Alexandroupolis
Full PDF | ![]()
Haematological malignancies which occur in a temperate climate also occur in tropical and subtropical countries.1-4 However, there are notable differences in prevalence and epidemiology between tropical and non-tropical regions:
plasma cell dyscrasias: The age-adjusted incidence rates of myeloma are 9.9/100000 in black Americans, 4.3/100000 in white Americans and 3.8/100000 in Asian Americans, i.e. myeloma is two times more common in blacks than whites. Also, MGUS is twice more common in blacks than whites, however, there is no difference in the rate of progression to myeloma (1% per year in both races). The high incidence of plasma cell dyscrasias in black Africans and African-Americans may be genetically determined, reflecting the higher normal values of serum immunoglobulins in blacks than whites. The diagnosis of myeloma is made at high frequencies in the Caribbean and in tropical Africa, where it commonly affects patients aged 30-39 years, and ~65% of patients are 40-60 years old. Plasmablastic myeloma and plasmacytomas with blastic morphology are not uncommon among black Africans and African-Americans and often the disease is aggressive.3,5-8 The definition of plasmablast (according to Greipp) is based on the following characteristics: large round nucleus (>10 μm) or large nucleolus (>2 μm), fine reticular (immature) chromatin with no (or minimal) densities, high nuclear:cytoplasmic ratio (i.e. cytoplasm <1/2 nuclear diametre), and absent (or minimal) clear perinuclear area (hof).
Chronic lymphocytic leukaemia (CLL) is rare throughout the Indian subcontinent, East Asia and even more rare in the Far East (China and Japan). CLL occurs in tropical Africa with equal numbers of men and women affected. About half the patients are aged ≤45 years; in these younger adults CLL is associated with low socioeconomic status.3,9-11 By contrast, CLL is the most common leukaemia in adults in Western countries, with male predominance and average age at diagnosis 72 years. The fact that CLL is 10-20 times commoner in Western countries than Asia, suggests that genetic factors (genetic polymorphisms?), environmental factors, or both influence susceptibility to the disease.
AIDS-related lymphomas (ARL) represent a major health problem in sub-Saharan Africa. Systemic ARL is ~70% DLΒCL and ~30% Burkitt’s lymphoma. The bone marrow is frequently involved in HIV-associated DLBCL, Burkitt’s lymphoma, Hodgkin’s lymphoma, and Castleman’s disease. The incidence of B-cell precursor ALL is also increased.3,12,13
Immunoproliferative small intestinal disease (IPSID). This condition occurs at high incidence in children and young adults in North Africa, Middle East, eastern Mediterranean (e.g. Israel), South-East Asia, India, and sub-Saharan Africa. Sporadic cases have been reported in South, Central and North America, and Europe. It is associated with low socioeconomic status and recurrent or chronic enteric infections (Campylobacter). There is steatorrhoea, malabsorption and weight loss due to infiltration of small intestine (jejunum) mucosa and mesenteric lymph nodes by plasma cells and plasmacytoid lymphocytes. The proliferating cells are IgA-producing B-cells, which synthesize defective immunoglobulin α heavy chains (IPSID is also known as α heavy chain disease). Serum protein electrophoresis is characteristic (in ~70% the serum or urine contain α heavy chains; in ~30% α chains are produced but not secreted). The bone marrow may show empty iron stores and megaloblastic change because the disease is complicated in 40% by malabsorption of iron, folate, or vitamin B12; a mild increase in plasma cells may be seen but bone marrow infiltration is uncommon. IPSID may progress to high-grade immunoblastic lymphoma rich in plasma cells (Mediterranean lymphoma).3,14,15
Tropical splenic marginal zone lymphoma with villous lymphocytes (T-SLVL) is more common in parts of Africa where malaria is hyperendemic and hyper-reactive malarial splenomegaly (HMS) has high frequency. In contrast to the western-type SLVL, T-SLVL occurs more often in young women. It is possible that HMS may progress to T-SLVL.16-18
Hodgkin’s lymphoma (HL): There are four epidemiological patterns: Pattern I: there are high incidences of HL in childhood in Central and South America, North Africa, western Asia and sub-Saharan Africa; there is a predominance of mixed cellularity (MC) and lymphocyte depletion (LD) histological subtypes; 50-70% are EBV (+). Pattern III: in developed countries, there is a peak of incidence in young adults (20-34 years), in whom nodular sclerosis (NS) is the predominant type and a second peak of HL in middle age or latter; 20-30% are EBV (+). Pattern II: this is intermediate between patterns I and III, and is found in rural areas of developed countries including eastern Europe and southern USA. Pattern IV: HL has a low incidence in eastern Asia.3,19
Tropical haematological malignancies:
1. Endemic Burkitt’s lymphoma1-4,20-22
It occurs in children within the lymphoma belt of Africa (about 10o N to 10o S of the Equator), Papua New Guinea and parts of the Amazon basin and Malaysia, where malaria is holoendemic (a holoendemic disease is one for which a high prevalent level of infection begins early in life and affects most of the child population, leading to a state of equilibrium such that the adult population shows evidence of the disease much less commonly than do children). Burkitt’s lymphoma (BL) is the most common childhood tumour (>50%) in Nigeria, Uganda, and Papua New Guinea (‘tropical lymphoma’).
The geographical distribution of the tumour in high incidence is mainly controlled by two climatic paramaters, temperature and humidity (Figure 1).

Figure 1. Climatic dependence of endemic Burkitt’s lymphoma.
In tropical Africa as well as other continents the distribution of Burkitt’s tumour is roughly the same as that of malaria.
Burkitt᾽s lymphoma is associated with EBV infection. EBV infection typically occurs during one of two distinct time periods: the first wave of infection occurs during early childhood, before the age of 5 years (tropical countries), and the second wave occurs during the second and third decades of life (western countries).
There are three epidemiological patterns of BL: (I) BL is endemic in children in tropical Africa and Papua New Guinea, where annual incidence is 8-12/100000, with a peak at 4-9 years of age (endemic, type A, or African Burkitt’s lymphoma). Children affected with the disease often also have chronic malaria which is believed to have reduced resistance to the Epstein-Barr virus and allowed it to cause malignant transformation; (II) intermediate incidence of 1-2/100000/year is found in children in North Africa, western Asia and South America (intermediate endemicity); (III) occasional cases, <0.1/100000/year, in the Western world (so-called type B, sporadic, non-endemic, western or American BL).3 Another variant of BL is immunodeficiency-associated BL, particularly occurring in association with AIDS (~30% of AIDS-related lymphomas). Notably, Burkitt’s lymphoma can be the initial manifestation of AIDS.
By morphology or flow immunophenotype, it is almost impossible to differentiate these three clinical variants. Immunodeficiency-associated Burkitt’s lymphoma may demonstrate more plasmacytic appearance or more pleomorphism, but these features are not specific. Table 1 shows characteristics of endemic, sporadic, and HIV-associated BL.
Causation (relationship with EBV and malaria):
Pattern I: ΕΒV 100% and P. falciparum recurrent infections (the majority of children in developing countries are infected by the EBV before the age of 1 year).
Pattern II: ΕΒV 80-100% (in areas of intermediate endemicity there are high rates of EBV transmission in early childhood before the age of 5 years), but no or low malaria transmission.
Pattern III: ΕΒV 20% (20% or less of sporadic cases of BL are associated with EBV) and no malaria.
HIV-associated BL: EBV 25-40% (its incidence is not reduced by highly active anti-retroviral therapy/HAART) and no malaria.
The leukaemic variant of BL, Burkitt cell leukaemia (L3 subtype or mature B-cell acute lymphoblastic leukaemia), is morphologically characterised by large, homogenous blasts with cytoplasmic vacuoles (monomorphic). It is a common form of acute leukaemia in the tropics. In contrast, it is the most uncommon morphological type of ALL in the westerm countries (4%). Burkitt’s lymphoma and the L3 subtype of acute lymphoblastic leukaemia can be regarded as variants of a single disease. In endemic Burkitt’s lymphoma a leukaemic phase typically occurs as a late event and is associated with a poor prognosis. A leukaemic presentation is more common in non-endemic Burkitt’s lymphoma, in which it is compatible with cure of the disease as long as appropriate intensive chemotherapy is given. In AIDS-related Burkitt’s lymphoma, bone-marrow and peripheral-blood involvement are common and are associated with poor prognosis.
Features of Burkitt cells:22
The features considered are:
| Cytolological features | Burkitt cells |
| Cell size | Large and homogeneous |
| Nuclear chromatin | Dense but finely stippled and homogeneous |
| Nuclear shape | Regular-oval to round, no clefting, identation or folding |
| Nucleoli | Prominent; one or usually more vesicular |
| Amount of cytoplasm | Moderately abundant, surrounds the nucleus, no granules |
| Basophilia of cytoplasm | Very deep (due to high RNA content) |
| Cytoplasmic vacuolation | Often prominent in a majority of the cells |
Burkitt cell leukaemia is characteristically homogeneous in respect of every one of the seven morphological features listed, both from case to case, and in an individual case (for each of the features listed, occasional cells <10% may depart from the classical morphology!).
Tips:
– a high mitotic index (about 5%) is characteristic: in the bone marrow aspirates you should be able to find several mitotic figures (prophases, metaphases, anaphases, cytokineses). If you cannot find mitotic figures → the diagnosis of BL is questioned!
– the cytoplasmic vacuoles of Burkitt cells contain lipids. They are always PAS (-).
– all listed seven features are required for diagnosis of Burkitt cell leukaemia and differentiation from lymphoblasts of B-cell precursor ALL, other high-grade non-Hodgkin lymphoma (especially Burkitt-like high-grade B-cell lymphoma, double-hit or triple-hit lymphoma, plasmablastic lymphoma), blastic mantle-cell lymphoma (sometimes vacuoles are prominent), AML blasts (especially monoblastic leukaemia in which vacuoles may be striking), and non haematological tumours of childhood which commonly involve the bone marrow e.g. rhabdomyosarcoma in which vacuoles are typically prominent. Flow-cytometric immunophenotyping can help in differentiating BL from other aggressive B-cell lymphomas (Burkitt lymphoma cells are homogenously CD38 bright+, CD10 bright+, BCL 2-, and Ki-67 strongly +).23
Warning: the presence of vacuoles in immature cells is not enough to justify Burkitt cells! For example, the cells in Figure 2 have nuclear folding and identations and, therefore, are not morphologically consistent with Burkitt cells (this was a case of acute monocytic leukaemia).
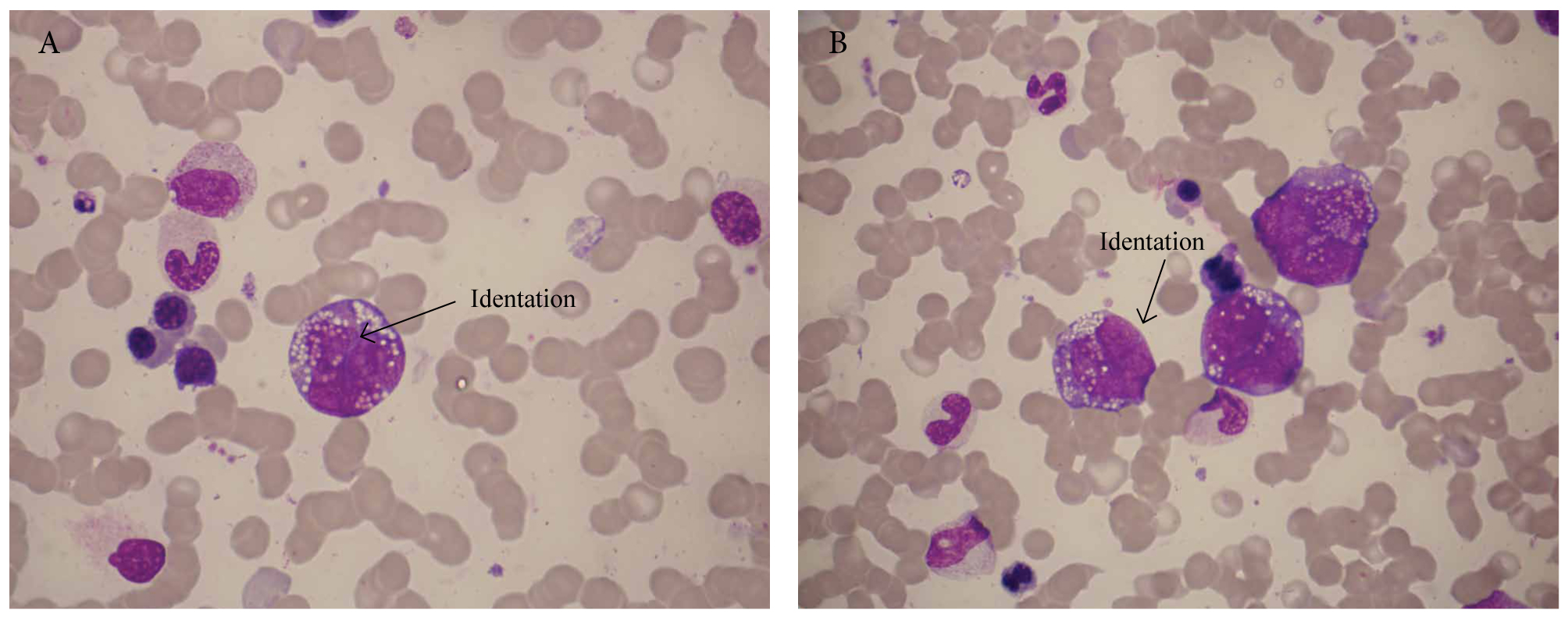
Figure 2. Acute monoblastic leukaemia with prominent cytoplasmic vacuoles.
Figure 3 is a typical example of Burkitt cells.

Figure 3. A 4-year-old girl from Jeddah Saudi Arabia was referred because of fever and persistent neutropenia. Blood tests showed WBC 1.4 ×109/l, Hb 10.3 g/dl, PLT 270×109/l, and ΜCV 90 fl. Bone marrow examination showed 63% infiltation by ΕΒV(+) Burkitt cell leukaemia (epidemiological pattern ΙΙ).
2. Adult T-cell leukaemia/lymphoma (ATL)2-4,24-26
HTLV-1, transmitted by sexual intercourse, through breast-feeding, by blood transfusion and i.v. drug use, is endemic in geographical clusters in Asia, Africa, the Americas, and Australia.
Seroprevalence is high (up to 30%) in South-West Japan. HTLV-1 is encountered sporadically in other parts of Japan and in Taiwan, and only occasionally in other Asian countries.
The largest pool of the virus is in sub-Saharan Africa: the highest rates of seropositivity are in the rainforests of West Central Africa (up to 15% has been reported in Congo, and 10% in Gabon and southern Cameroon); prevalence declines northwards into the savannah (3-6% in the savannah of Nigeria); rates are lower in coastal areas and further west as far as Senegal (1-2%); HTLV-1 has low endemicity in populations of East and Central Africa (~1%), except in the Seychelles, where overall frequency is 6.2%; in South Africa the frequency is 1-3.5%.
The virus is also endemic in populations of African descent in the Caribbean, Central America, southern USA, and Britain (3-6% among Jamaican adults living in the UK).
Clusters have been reported in Iran, Iraq, the Caucasus, central Australia (45% in indigenous communities!), Papua New Guinea, and the Solomon Islands.
Seroprevalence rises slowly with age, compatible with the slow rate of sexual transmission in endemic areas. HTLV-1 is spreading epidemically amongst i.v. drug users and men who have sex with men (MSM) in North and South America (Brazil) and western and eastern Europe especially Romania.
There is a spectrum of clinical syndromes in ATL: 5 clinical phases are recognised according to Shimoyama classification:26
asymptomatic ATL: some asymptomatic patients show a preleukaemic condition, diagnosed from the incidental observation of lymphocytosis with abnormal cells characterised by pleomorphism, multilobed nuclei (‘flower’ or ‘clover-leaf’ cells), or cytoplasmic vacuoles; pre-ATL is transient in about half of patients, but may persist and progress to ATL.
Note: all types of ATL are neoplasms and across the whole spectrum of ATL a T-cell clone is detected by analysis of gamma and beta genes by PCR or evidence of clonal HTLV-1 intergration, unlike HTLV-1 carriers (HTLV-1 carriers are positive for HTLV-1 Ab, but have no clonal T-cells).
chronic ATL (~20% of the African and Caribbean patients): skin lesions and mild lymphocytosis only, few circulating ATL cells with abnormal nuclear outline (small sized), and normal bone marrow. Prolonged course (the chronic type shows an indolent clinical course and the patients survive for several years even without chemotherapy but some patients have unfavourable prognostic factors).
smouldering ATL (5%): mild skin rashes; normal WBC count (no lymphocytosis); low count of circulating ATL cells (but >5% of WBC); remains stable for many years (very slow disease progression).
lymphoma type ATL (20%): no circulating ΑΤL cells (<1%); it is clinically like NHL (diffuse and polymophous lymphoma consisting of small, larger, and large cells), without the leukaemic manifestations; high serum lactate dehydrogenase (LDH).
Note: The histological features of lymph nodes in ATL may be indistinguishable from those of other peripheral T-cell lymphomas; HTLV-1 serology is needed for diagnosis.
acute ATL (~50%): lymphadenopathy, hepatosplenomegaly, hypercalcaemia, rash, pulmonary infiltration and osteolyses. Acute ATL is a systemic disease: there is always blood involvement i.e. ATL cells in the blood film (at least 5% circulating abnormal T-cells is required to diagnose ATL) and in most patients generalised lymphadenopathy, hepatosplenomegaly, high LDH (characteristic), and frequently (>50%) rash (biopsy-proven infiltration), systemic symptoms, hypercalcaemia (with or without bone lesions), and eosinophilia (rule out Strongyloides!). Anaemia and thrombocytopenia are often absent. The WBC count is usually raised, 30-130×109/l, with a predominance of the characteristic abnormal lymphocytes (particularly in the Japanese patients who often have more pronounced leukaemic blood picture than the Caribbean patients). The bone marrow shows infiltration, but less than would be expected from the leukaemic blood picture. Lungs, liver, GI tract, peritoneum, and CNS may be involved. Karyotypic lesions are mainly seen in the acute-type ATL (they are rare in chronic and smouldering types). The acute type progresses rapidly (usually survival is <1 year).
T-cell function is defective: S. stercoralis hyperinfection and other opportunistic infections e.g. PCP are associated with HTLV-1 infection at any stage of the ATL spectrum! It is common practice to give a course of albendazole to all ATL patients, irrespective of their eosinophil counts.27
The lifetime risk of developing ATL is about 5% in subjects infected by HTLV-1 before 20 years of age. The incubation period is decades: the mean age of onset is 40-45 years, but it can be seen rarely in adolescence; women are affected more often than men in Africa and the Americas. In contrast, in Japan the mean age at diagnosis is nearly 60 years and men are affected more often than women. It is possible that HIV co-infection may accelerate progression to ATL in tropical Africa.
Features of ATL cells:3,29
These are circulating abnormal lymphoid cells with striking polymorphism with small, medium-sized, and large cells (the cells vary considerably in size from that of a small lymphocyte to a large mononuclear cell >14 μm) and characteristic nuclear appearance: abnormal polylobed nucleus (the nuclear outline has been described variously as convoluted, ‘flower like’, polypetaloid, or ‘clover leafed’. The nuclear chromatin is relatively homogeneous and clumped or condensed. Nucleoli are uncommon and when present are small. The cytoplasm is agranular and basophilic (Figure 4). Some cells contain small vacuoles in the cytoplasm (necklace-like). In addition to polylobed cells, some blastic cells with basophilic cytoplasm (immunoblasts) are almost always seen in blood films of ATL. The cells are positive for acid phosphatase (Golgi-type) and α-naphthyl acetate esterase (ΑΝΑΕ). The white cell count ranges widely and the percentage of abnormal cells ranges from 10-80%.

Figure 4. A-D. A 42-year-old woman, originally from Antigua (Antilles), with acute-type ATL who presented with rash, serum calcium 14 mg/dl, and high LDH (1800 U/l). Findings include striking polymorphism, flower cells, and clover-leaf cells; also note rbc morphology with numerous target cells (Hb C carrier).
Occasionally it is difficult to distinguish the cells from those seen in Sézary’s syndrome (Figure 5). Sézary syndrome is a rare variant with an aggressive clinical behaviour, presenting with erythroderma, generalised lymphadenopathy and leukaemic blood involvement. The neoplastic T-cells are thought to be CD4+ TCRaß+ memory cells with skin-homing properties and a Th2 phenotype. The eosinophil count is frequently elevated in both ATL and Sézary syndrome (but concurrent helminthic infection e.g. strongyloidiasis should always be ruled out in such cases). There are also instances in which T-prolymphocytic leukaemia (T-PLL) may be confused with ATL because the prominent nucleolus normally seen in T-PLL may be indistinct and the nuclear outline may be irregular (cerebriform variant T-PLL). The cells are CD4+/CD7- and, unlike other T-cell disorders, typically express the receptor for IL-2α (CD25) but this may also be seen in other chronic T-cell leukaemias/lymphomas. Rarely, ATL may be ‘double-negative’ i.e. CD3+/CD4-/CD8-.28,30 The cells originate from helper T-cells with functional suppressor activity (T-regs?).

Figure 5. A-D. A 50-year-old woman, originally from Jamaica, with acute-type ATL presenting with rash, lymphadenopathy, and refractory severe hypercalcaemia. This was a case of cerebriform variant ATL.
Differential diagnosis of ΑΤL cells:
Sézary’s syndrome, cerebriform T-PLL, T-NHL (particularly AITL), T-cell ALL
Similarities:
T-PLL → 65% CD4+, 5% cerebriform, 25% skin involvement but T-PLL is typically CD7+ and TCL1+.
ATL → CD4+, >50% skin involvement, may be cerebriform, but ATL is typically CD7- and CD25 strongly +.
ATL patients are always HTLV-1 Ab (+). HTLV-1 Ab is a very useful test in the diffential diagnosis of T-cell lymphocytosis. Consider HTLV-1 serology in ‘atypical’ unclassified CD4+ lymphocytosis with clonal population on TCR PCR, in cases of Τ-cell lymphoma with hypercalcaemia, in cases of atypical skin rash with circulating cells with convoluted or cerebriform nuclei, and in cases of bizarre looking lymphomas with Hodgkin-like cells, particularly in patients orignating fom endemic areas (sub-Saharan Africa, Caribbean).
Tips:
ATL is a leukaemia with polymorphic polylobed cells.
ATL cells have two morphological characteristics: pronounced polymorphism and irregular nuclear outline (many cases do not show the classic flower cells).
Where can we find circulating cerebriform CD4+ lymphocytes?
– Sézary’s syndrome (90-100% skin involvement, lymphadenopathy+)
– cerebriform T-PLL (25% skin involvement, lymphadenopathy -/+))
– ATL (50-70% skin involvement, lymphadenopathy+)
– actinic reticuloid (a benign dermatological disorder with Sézary-like cells)
– rarely, circulating cerebriform T-cells may be seen in T-ALL (mature-T subset) and AITL (CD4+CD10+ CD200+CD7-).31
Remember: there is a spectrum of clinical syndromes in ATL.
References
- Peters W, Gilles HM. A colour atlas of tropical medicine and parasitology. 2nd ed. London: Wolfe; c1981.
- Peters W, Pasvol G. A colour atlas of tropical medicine and parasitology. 6th ed. London: Mosby Elsevier; c2007.
- Cook GC, Zumla AI. Manson’s tropical disease. 22nd ed. London: Εlsevier Saunders; c2008.
- Farrar J, Hotez P, Junghanss T, Kang G, Lalloo D, White N. Manson’s tropical diseases. 23rd ed. London: Εlsevier Saunders; c2014.
- Kyle RA, Rajkumar SV. Multiple myeloma. Blood 2008 Mar;111(6):2962-72.
- Kyle RA, Rajkumar SV. Epidemiology of the plasma cell disorders. Best Pract Res Clin Haematol. 2007 Dec;20(4):637-64.
- Landgren O, Kyle RA, Pfeiffer RM, et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: a prospective study. Blood. 2009 May;113(22):5412-7.
- Blattner WA, Jacobson RJ, Shulman G. Multiple myeloma in South African blacks. Lancet. 1979 Apr;1(8122):928-9.
- Fleming AF. Leukaemias in Africa. Leukemia. 1993 August;7 Suppl 2:S138-141.
- Fleming AF. Chronic lymphocytic leukaemia in tropical Africa: a review. Leuk Lymphoma. 1990;1(3-4):169-73.
- Shih L-Y, Liang D-C. Non-Hodgkins lymphomas in Asia. Hematol Oncol Clin North Am. 1991 Oct;5(5):983-1001.
- Liapis K, Clear A, Owen A, Coutinho R, Greaves P, Lee AM, et al. The microenvironment of AIDS-related diffuse large-B-cell lymphoma provides insight into the pathophysiology and indicates possible therapeutic strategies. Blood. 2013 Jul;122(3):424-33.
- Bower M. Acquired immunodeficiency syndrome-related systemic non-Hodgkin’s lymphoma. Br J Haematol. 2001 Mar;112(4):863-73.
- Bouchier IAD, Allan RN, Hodgson HJF. Gastroenterology: Clinical Science and Practice. 2nd ed. London: W. B. Saunders; c1993.
- Ghoshal UC, Chetri K, Banerjee PK, Choudhuri G, Pal BB, Dabadghao S, et al. Is immunoproliferative small intestinal disease uncommon in India? Trop Gastroenterol. 2001 January-Mar;229(1):14-7.
- Bedu-Addo G, Bates Ι. Causes of massive tropical splenomegaly in Ghana. Lancet. 2002 Aug;360(933):449-54.
- Bates I, Bevan DH, Rutherford TR, Bedu-Addo G. Use of immunoglobulin gene rearrangements to show clonal lymphoproliferation in hyper-reactive malarial splenomegaly. Lancet. 1991 Mar;337(8740):505-7.
- Bates I, Bedu-Addo G, Rutherford TR, Bevan DH. Circulating villous lymphocytes – a link between hyperreactive malarial splenomegaly and splenic lymphoma. Trans R Soc Trop Med Hyg. 1997 Mar-Apr;91(2):171-4.
- Glaser SL. Hodgkin’s disease in black populations: a review of the epidemiologic literature. Semin Hematol. 1990 Dec;17(6):643-59.
- van den Bosch CA. Is endemic Burkitt’s lymphoma an alliance between three infections and a tumour promoter? Lancet Oncol. 2004 Dec;5(12):738-46.
- Burkitt D. The discovery of Burkitt’s lymphoma. Cancer. 1983 May;51(10): 1777-86.
- Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DAG, Gralnick HR, Sultan C. Proposals for the classification of the acute leukaemias. (FAB Cooperative Group). Br J Haematol. 1976 Aug;33(4):451-8.
- Tsagarakis NJ, Papadhimitriou SI, Pavlidis D, Liapis K, Gortzolidis G, Kostopoulos IV, et al. Contribution of immunophenotype to the investigation and differential diagnosis of Burkitt lymphoma, double-hit high-grade B-cell lymphoma, and single-hit MYC-rearranged diffuse large B-cell lymphoma. Cytometry B Clin Cytom. 2020 Jun 4 [Online ahead of print].
- Manns A, Hisada M, La Grenade L. Human T-lymphotropic virus type I infection. Lancet. 1999 Jun;353(9168):1951-8.
- Human T-lymphotropic virus 1: recent knowledge about an ancient infection. Lancet Infect Dis. 2007 Apr;7(4):266-81.
- Shimoyama M. Diagnostic criteria and classification of clinical subtypes of adult T-cell leukaemia-lymphoma. A report from the Lymphoma Study Group (1984-87). Br J Haematol. 1991 Nov;79(3):428-37.
- Kwon R, Duffield AS. HTLV-1-associated adult T-cell leukemia/lymphoma with disseminated strongyloidiasis. Blood. 2019 Jun;133(24):2623.
- Khanlari M, Ramos JC, Sanchez SP, Cho-Vega JH, Amador A, Campuzano-Zuluaga G, et al. Adult T-cell leukemia/lymphoma can be indistinguishable from other more common T-cell lymphomas. The University of Miami experience with a large cohort of cases. Mod Pathol. 2018 Jul;31(7):1046-63.
- Bennett JM, Catovsky D, Daniel MT, Flandrin G, Galton DA, Gralnick HR, Sultan C. Proposals for the classification of chronic (mature) B and T lymphoid leukaemias. French-American-British (FAB) Cooperative Group. J Clin Pathol. 1989 Jun;42(6):567-84.
- Liapis K, Tsagarakis NJ, Panitsas F, Taparkou A, Liapis I, Roubakis C, et al. Causes of double-negative T-cell lymphocytosis in children and adults. J Clin Pathol.2020 Jul;73(7):431-8.
- Liapis K, Paterakis G. Circulating angioimmunoblastic T-cell lymphoma cells. Blood. 2020 Apr;135(18):1607.