Review Article
Haema 2019; 10(1):29-38
Photis Beris
MD, Scientific director of French speaking region of Hematology department Unilabs, Switzerland. Former Professor of Hematology Geneva University Hospital, Geneva School of Medicine and former Professor of Hematology, University of Athens, School of Medicine.
Full PDF | ![]()
Abstract
Immunologic reactions were the first obstacle and risk, related to transfusions. They were resolved by the discovery of blood groups and the progress in immunohematology. Transmission of infectious agents became thereafter a 2nd problem with a lot of victims. Screening tests and viro-inactivation techniques have made transfusion safe (actual residual risk almost zero in Switzerland). Iron overload is still a problem in transfusion-dependent patients. What can we do to fight against iron overload? 1. Correct anemia (HSCT) or improve it (some forms of MDS with inefficient erythropoiesis, thalassemia intermedia) using anti-GDF11 drugs. 2. Try to correct iron hyper absorption which is associated with low levels of hepcidin. 3. In case of sickle cell anemia correction of iron overload by erythrocytapheresis. 4. Sometimes (in case of low-risk MDS anemia), chelation removes the toxic effect of iron on erythropoiesis and transforms transfusion dependent anemia in transfusion-independent. 5. Chelation of iron by available drugs: the most effective way which applies to all cases. Increased concentrations of TGFβ ligand superfamily, including GDF11, are associated with inefficient erythropoiesis in MDS and other forms of anemia characterized by inefficient erythropoiesis. Luspatercept and Sotatercept, (hybrid molecules, fusion proteins, composed of the extracellular domain of the human activin IIA receptor and the Fc part of IgG1), are competitors of the activins and morphogenic proteins of the bone and thus block the signal transduction via the SMAD path. For anemic patients with inefficient erythropoiesis, the use of these inhibitors of the TGFβ pathway could improve Hb levels and decrease transfusion requirements. The results of Phase 2 studies go in this direction.
Key words: Transfusion related reactions, Iron overload, Anti-GDF11 drugs, Erythrocytapheresis, Iron chelation, Anemia in MDS
Corresponding author: Photis Beris, 51 avenue Blanc, 1202 Genève-CH, e-mail: photis.beris@unilabs.com
INTRODUCTION
Chronic transfusion allows to avoid complications in patients suffering from sickle cell anemia, to survive in patients with beta thalassemia major, or with Diamond-Blakfan anemia or with a form of CDA (Congenital Dyserythropoietic Anemia), or even suffering from anemia secondary to MDS, myelofibrosis or aplastic anemia. However chronic transfusion presents risks of serious, sometimes mortal complications. Because human beings do not possess effective mechanisms to get rid of iron, chronic transfusion leads to iron overload responsible of lesions in parenchymal organs and or in dysfunctions of endocrine glands.
Pathophysiology of iron overload in chronically transfused patients may be summarized as follows: In a first step transfused erythrocytes are phagocytized by RES (reticulo-endothelial-system). The iron is then liberated from heme and cumulates in the cytoplasm. Macrophages release iron excess in the plasma where iron is bound to transferrin. This progressively leads to increased saturation of serum transferrin. Hepatocytes are another site of iron storage. Chronic transfusion exceeds loading capacity of iron in macrophages and hepatocytes. A huge amount of iron is released in the plasma which exceeds transferrin’s capacity to transport iron and thus we have production of NTBI (non-transferrin bound iron), which is the toxic form of iron to cells via the formation of ROS (reactive oxygen species).1
Figure 1 schematically represents organ lesions secondary to iron overload in chronically transfused patients (From Reference 1).

Figure 1. Schematically representation of organ lesions secondary to iron overload in chronically transfused patients (From reference 1).
However, transfusion is not the only source of excessive iron input. We must remind here the role of hepcidin and particularly the role of erythroferrone in certain forms of anemia characterized by accelerated and ineffective erythropoiesis.
Figures 2 and 3 illustrate the role of these two proteins in iron absorption.
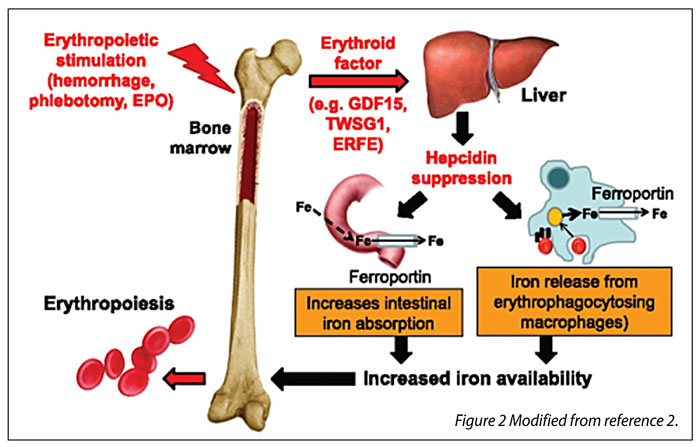
Figure 2. Role of erythroferrone and hepcidin in iron absorption in normal situation.

Figure 3. Role of erythroferrone and hepcidin in iron absorption in hemolysis and ineffective erythropoiesis.
In brief erythropoietic stimulation leads to production of erythroid factors among which is erythroferrone (ERFE). These factors suppress hepcidin production leading to increased iron absorption from the intestine and to iron release from macrophages. The increased iron availability covers iron needs for accelerated erythropoiesis (Figure 2). In case of ineffective erythropoiesis, the increased iron absorption and the iron brought by transfusions lead to iron overload (Figure 3, Reference 2).
In our laboratory, we have studied hepcidin levels in autoimmune hemolytic anemia induced in mouse. In Figure 4 it is clearly seen that induction of autoimmune anemia in mice leads to significantly decrease of hepcidin levels in day 4 (Figure 5) after administration of anti-erythrocytic antibody.3 We believe that any form of accelerated erythropoiesis leads at a collapse of hepcidin and this because of the hyper-production of erythroferrone.

Figure 4. Induction of immune hemolytic anemia by using antierythrocytic antibody in mice.

Figure 5. Induction of autoimmune anemia in mice leads to significantly decrease of hepcidin levels in day 4.
Let us see now the risks of chronic transfusion in different clinical settings:
1. CHRONIC TRANSFUSION IN CASE OF MDS: WHERE ARE THE RISKS
Iron overload in case of MDS starts before the patients become transfusion-dependent because, as we saw before, ineffective erythropoiesis inhibits production of hepcidin.4 However chronic transfusion is the main and most important source of iron overload in anemic MDS patients: For example, if a patient receives 4 units/month this corresponds in 100 units per two years, which means 20 gr of iron.5 Clinical studies have shown that iron overload in MDS decreases survival in patients with low risk MDS.6 Furthermore, iron overload leads at endothelial cell dysfunction which aggravates arteriosclerosis.7 Iron overload because of oxidative stress of ROS favors the evolution to acute leukemia.8 It was recently shown that iron overload deteriorates erythropoiesis which means aggravates anemia.9
Figures 6 and 7 illustrate the possible role for iron overload in the pathophysiology of MDS (Figure 6 from Reference 5; Figure 7 from Reference 10)

Figure 6. Iron overload because of oxidative stress of ROS favors the evolution to acute leukemia (From reference 5).
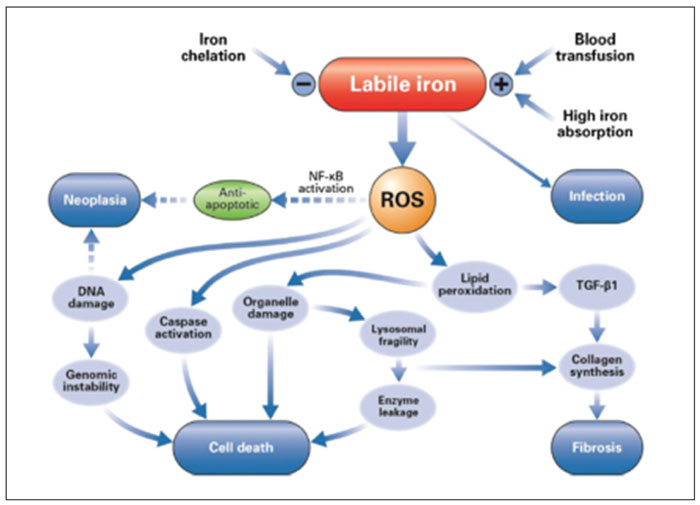
Figure 7. Possible role for iron overload in the pathophysiology of MDS (from reference 10).
2. THE ROLE OF IRON OVERLOAD IN HSCT IN CASE OF THALASSEMIA, MDS BUT ALSO IN AL
In case of iron overload in patients submitted to HSCT (Haematopoietic Stem Cell Transplantation), elevated liver iron concentration (LIC) is associated and, this significantly, with reduced overall survival post HSCT.11 Furthermore, there is an increased risk of infectious complications particularly by fungi. If LPI (labile plasma iron), is detectable before transplantation, then patients are at high risk for mortality non-linked to relapse of the malignant hematologic disease.12 After transplantation, treatment by phlebotomy or by chelators improves hepatic function and decreases GvHD.13 It is highly suspected that chelation before transplantation may be extremely important but this should be proved by prospective randomized studies before routine application in the clinic.10
What can we do to fight against iron overload in such settings?
- If possible correct anemia by HSCT or improve it in certain forms of MDS and ineffective erythropoiesis or in thalassemia intermedia, by using the new category of drugs with anti GDF11 properties (see later in this text).
- Try to decrease iron hyperabsorption linked with low hepcidin levels (see also Figure 8).
- In case of sickle cell anemia, anemia and iron overload can be corrected by erythrocytapheresis (see also Figure 9).
- It was recently shown, in cases of low risk MDS, that iron chelation reduces toxic effect of iron to erythropoiesis transforming thus the patients in transfusion independent ones or reducing transfusion needs (Table 1).
- The most important: Iron chelation by drugs

Figure 8. Transfusion suppresses erythropoiesis and increases hepcidin in adult patients with β-thalassemia major. This leads to decreased iron absorption which explains why pre- and post-transfusion ferritin levels remain almost the same (From reference 17).

Figure 9. Serum ferritin levels in 6 patients with HbSS disease, transfusion-dependent and treated by erythrocytapheresis.18
It is possible that suppression of oxidative stress improves hemopoiesis by slowing down malignant clonal genetic evolution and by offering a better support by the bone marrow stroma.5
In β-thalassemia major, hepcidin levels are simultaneously associated with erythropoiesis and iron loading pre- and posttransfusion. Transfusion improves anemia, suppressing erythropoiesis and in turn increasing hepcidin in patients with β-thalassemia major. This was showen by Pasricha et al in a longitudinal study published in Blood in 2013.17 It is clearly seen in Figure 8 that transfusion leads to decreased production of erythroid factors like GDF15 (Fig 8C), with as a consequence an increase of hepcidin in the post transfusion period (Fig 8G) and this explains the stability of ferritin levels (Fig 8E).
Erythrocytapheresis in case of iron overload in Sickle cell anemia combines phlebotomy with transfusion. In fact, reduction of HbS concentration to 25-30% allows to « pump » iron from the iron stores of the patient, by producing new Hb S and this up to 50-55%, value when a new erythrocytapheresis will be done (in general every 4 to 8 weeks).
The main indications in sickle cell anemia are CVA (cerebrovascular accidents), acute chest syndrome, pulmonary hypertension. This therapeutic strategy is very effective against iron overload in this setting (Figure 9). In our hands it leads to iron deficiency anemia! However, the principal obstacle for a routine use is the vein access to perform the procedure.18-21
Treatment of iron overload in patients with MDS: indications and therapeutic scheme
Treatment by chelators should started in MDS patients with low or intermediate risk according to IPSS-R (RA, RARS or isolated 5q-), in patients transfusion-dependent, with ferritin >1000 μg/l (= after 25 units). However significant association between survival and ferritin was found when a ferritin level of ≥500μg/l was considered. In these patients it is possible that chelation treatment by improving ineffective erythropoiesis, improves survival.22 Chelation should also start in patients with MDS candidates for allogeneic HSCT.
Concerning choice of chelator, the most frequently used is deferasirox in its new formulation FCT (film-coated tablet)23 and in the following dosages:
→15mg/kg (FCT) if transfusion needs are <2 Units/month;
→20mg/kg (FCT) if transfusion needs are intermediates
→25-30mg/kg (FCT) if transfusion needs are 2 or more units /month.5
Treatment of iron overload in patients with Thalassemia
The following Table 2 gives thresholds values to evaluate iron overload in patients with thalassemia.1,10
In thalassemia chelation should start at mild iron overload state. It is considered effective if the patients stays at this state and does not progress to moderate or severe iron overloaded state (Table 2). However, if this is the case then intensive iron chelation is recommended (see below).
Intensive iron chelation
Is indicated in thalassemic children (patients) with moderate or severe iron overload, leading to heart failure. In the literature we find many such cases successfully treated with intensive iron chelation.24 This is also valid for similar situations in patients with MDS.25
The applied regimen is deferoxamine (DFO) 30-50mg/kg/d 5x/week associated to deferasirox 15-20mg/kg/d or to deferiprone 50-75mg/kg/d divided in 3 doses.
The evaluation of treatment is done by measuring T2*, ferritin levels and pro-BNT levels and by performing cardiac ultrasounds.25,26
Measurement of NTBI and LPI (=labile plasma iron)
LPI represents the most toxic component of NTBI and is an active redox factor. It penetrates cellular membranes and destroys cells and tissues by the production of ROS. It was demonstrated that LPI has a significant relation with LIC and it provides a supplementary indication of treatment response by iron chelators as this is shown in the next Table 3.
For the time being there is no standardized method for the quantification of serum non-transferrin bound iron and labile plasma iron, in iron overload patients. We quote here the comments of the 2nd international round robin for the quantification of NTBI & LPI published in Haematologica:29 “Absolute levels differed considerably between assays and were lower for LPI than for NTBI. Four assays also reported negative values. Assays were reproducible with high between-sample and low within-sample variation. Assays correlated and correlations were highest within the group of NTBI assays and within that of LPI assays. Increased transferrin saturation but not ferritin, was a good indicator of the presence of forms of circulating NTBI. The possibility of using NTBI & LPI measures as clinical indicators of overt iron overload and/or of treatment efficacy would largely depend on the rigorous validation and standardization of assays”.
Chelators used in the clinical practice
In the following Table 4 we give some characteristics of the three approved chelators actually used in clinical practice.10,28
Improvement of anemia in certain forms of MDS with ineffective erythropoiesis and in patients with thalassemia intermedia by using drugs with anti GDF11 effect: inhibition of the TGFβ pathway
Increased concentrations of the ligands of the superfamily TGFβ including GDF11 are linked with ineffective erythropoiesis in some cases of MDS.
Luspatercept and Sotatercept, are hybrid molecules, representing fusion proteins, composed of the extracellular domain of the receptor IIA of human activin and the Fc part of IgG1. They are competitors of activins and of bone morphogenetic proteins blocking thus the transduction of signal via the SMAD pathway.30,31
Their stimulatory activity of erythropoiesis was found in women treated by Sotatercept for osteoporosis.32,33 The following Figure 10 illustrates how inhibition of the TGF-β family member GDF11stimulates the last phase of erythropoiesis. It offers also a Model of crosstalk between erythropoiesis and iron metabolism.2
Phase 2 studies in patients with lower-risk myelodysplastic syndromes as well as in thalassemia intermedia patients have already shown the effectiveness of these drugs in anemia and consequently in decreasing iron overload. Hb concentration may improve up to 25 g/l as is shown in the following Figure 11.31

Figure 10. Model of crosstalk between erythropoiesis and iron metabolism involving TGF-β family member GDF11. It is also shown that inhibition of the TGF-β family member GDF11stimulates the last phase of erythropoiesis (From reference 2).

Figure 11. Change in Hb concentration from baseline in patients with low transfusion burden in the base study (A) and patients with low transfusion burden who were treated across both base and extension studies.31
In fact 32 of 51 patients (=63%) by receiving a dose of Luspatercept of 0.75-1.75 mg/kg/ weeks achieved an HI-E (Hematological improvement-erythroid).
Severe side effects were: muscle pain in 2% of patients and deterioration of physical condition in 2% of the treated patients. Basal EPO levels were predictive of response. Patients with acquired sideroblastic anemia and /or SF3B1 mutation respond better (77%). A phase 3 study is going on in case of lower risk MDS (NCT02631070).
The future: hepcidin agonists as therapeutic tools34
We saw earlier that hepcidin is the key factor in iron metabolism. Decreased hepcidin levels characterized all iron overload disorders. It is thus reasonable to prevent iron overload and improve erythropoiesis by creating drugs that either act as hepcidin agonists or stimulators of hepcidin production or even acting as ferroportin inhibitors.34 A recent paper in Blood provides the list of these drugs as well as the advance of clinical studies.33 These drugs will certainly change treatment of hereditary hemochromatosis. However, they are expected to be also beneficial, for the management of iron loading anemias characterized by low hepcidin levels because of hyper production of erythroferrone. These drugs may also complement other novel treatments mentioned before, that target ligands of the transforming growth factor-β family.35
CONCLUSIONS
Immunologic reactions were the first obstacle and mortal risk linked with transfusion. The discovery of blood groups and progress in immunohematology allowed to completely resolving them.
Transmission of infectious agents became the next major problem creating many victims. Serologic screening tests, diagnostic tests based in recombinant DNA technology as well as virus-inactivation techniques, allowed transfusion to become safe with a residual transmission risk close to zero in Europe, USA and some other countries.
Iron overload however continues to be a problem in chronically transfused patients.
If iron chelation by the new chelators, given orally, brings an acceptable solution to the problem, understanding iron metabolism in all its detail, will allow to arrive to an equilibrium in iron metabolism more easily, and in certain patients will even improve anemia with possibility to transform them transfusion-independent.
Such an example is given by findings, indicating increased concentrations of the ligands of the superfamily of TGFβ, including GDF11 in some anemic iron overloaded patients. These cases are characterized by ineffective erythropoiesis and belong mainly to low/intermediate-1 risk MDS patients as well as to thalassemia patients.
Luspatercept and Sotatercept are competitors of activin and of bone morphogenetic proteins inhibiting thus transduction of signal via the SMAD pathway.
For patients with anemia secondary to ineffective erythropoiesis, the use of these inhibitors of the TGFβ pathway, could improve Hb levels decreasing thus transfusional needs. Preliminary results of phase 2 studies, go in that direction.
Conflict of Interest
None.
REFERENCES
1. Quinn CT, St Pierre. TG MRI measurements of iron load in transfusion-dependent patients: implementation, challenges and pitfalls. Pediatr Blood Cancer. 2016 May;63(5):773-80.
2. Shenoy N, Vallumsertia N, Rachmilewitz E, Verma A, Ginzburg Y. Impact of iron overload and potential benefit from iron chelation in low-risk myelodysplastic syndrome. Blood. 2014 Aug;124(6):873-81.
3. Tchou I, Izui S, Beris P (oral communication, unpublished data, 2011).
4. Santini V, Girelli D, Sanna A, Martinelli N, Duca L, Campostrini N, et al. Hepcidin levels and their determinants in different types of myelodysplastic syndromes. PLoS One. 2011;6(8):e23109.
5. Gattermann N. Iron overload in myelodysplastic syndromes (MDS). Int J Hematol. 2018 Jan;107(1):55-63.
6. Malcovati L, Della Porta MG, Strupp C, Ambaglio I, Kuendgen A, Nachtkamp K, et al. Impact of the degree of anemia on the outcome of patients with myelodysplastic syndrome and its integration into the WHO classification-based Prognostic Scoring System (WPSS). Haematologica. 2011 Oct; 96(10):1433-40.
7. Vinci F, Muckenthaler MU, Da Silva MC, Balla G, Balla J, Jeney V. Atherogenesis and iron: from epidemiology to cellular level. Front Pharmacol 2014 May;5:94.
8. Harbort CJ, Soeiro-Pereira PV, von Bernurth H, Kaindl AM, Costa-Carvalho BT, Condino-Neto A, et al. Neutrophil oxidative burst activates ATM to regulate cytokine production and apoptosis. Blood. 2015 Dec;126(26):2842-51
9. Taoka K, Kumano K, Nakamura F, Hosoi M, Goyama S, Imai Y, et al. The effect of iron overload and chelation on erythroid differentiation. Int J Hematol. 2012 Feb;95(2):149-59.
10. Porter JB, de Witte T, Cappellini MD, Gattermann N. New insights into transfusion-related iron toxicity: implications for the oncologist. Crit Rev Oncol Hematol. 2016 Mar;99:261-71.
11. Leitch HA, Fibach E, Rachmilewitz E. Toxicity of iron overload and iron overload reduction in the setting of hematopoietic stem cell transplantation for hematologic malignancies. Crit Rev Oncol Hematol. 2017 May;113:156-70.
12. Wermke M, Eckoldt J, Götze KS, Klein SA, Bug G, de Wreede LC, et al. Enhanced labile plasma iron and outcome in acute myeloid leukaemia and myelodysplastic syndrome after allogeneic haemopoietic cell transplantation (ALLIVE): a prospective, multicentre, observational trial. Lancet Haematol. 2018 May;5(5):e201-10.
13. List AF, Baer MR, Steensma DP, Raza A, Esposito J, Martinez-Lopez N, et al. Deferasirox reduces serum ferritin and labile plasma iron in RBC transfusion-dependent patients with myelodysplastic syndrome. J Clin Oncol. 2012 Jun;30(17):2134-9
14. Gattermann N, Finelli C, Della Porta M, Fenaux P, Stadler M, Guerci-Bresler A, et al. Hematologic responses to deferasirox therapy in transfusion-dependent patients with myelodysplastic syndromes. Haematologica. 2012 Sep;97(9):1364-71.
15. Nolte F, Höchsmann B, Giagounidis A, Lübbert M, Platzbecker U, Haase D, et al. Results from a 1-year, open-label, single arm, multi-center trial evaluating the efficacy and safety of oral Deferasirox in patients diagnosed with low and int-1 risk myelodysplastic syndrome (MDS) and transfusion-dependent iron overload. Ann Hematol. 2013 Jan;92(2):191-8.
16. Angelucci E, Santini V, Di Tucci AA, Quaresmini G, Finelli C, Volpe A, et al. Deferasirox for transfusion-dependent patients with myelodysplastic syndromes: safety, efficacy, and beyond (GIMEMA MDS0306 Trial). Eur J Haematol. 2014 Jun;92(6):527-36.
17. Pasricha SR, Frazer DM, Bowden DK, Anderson GJ. Transfusion suppresses erythropoiesis and increases hepcidin in adult patients with β-thalassemia major: a longitudinal study. Blood. 2013 Jul;122(1):124-33.
18. Kim HC, Dugan NP, Silber JH, Martin MB, Schwartz E, Ohene-Frempong K, et al. Erythrocytapheresis therapy to reduce iron overload in chronically transfused patients with sickle cell disease. Blood. 1994 Feb;83(4):1136-42.
19. Dedeken L, Lê PQ, Rozen L, El Kenz H, Huybrechts S, Devalck C, et al. Automated RBC exchange compared to manual exchange transfusion for children with sickle cell disease is cost-effective and reduces iron overload. Transfusion. 2018 Jun;58(6):1356-62.
20. Delville M, Manceau S, Ait Abdallah N, Stolba J, Awad S, Damy T, et al. Arterio-venous fistula for automated red blood cells exchange in patients with sickle cell disease: complications and outcomes. Am J Hematol. 2017 Feb;92(2):136-40.
21. Lambert JF, Terrettaz M, Mach-Pascual S, Michel M, Beris Ph. Erythrocytapheresis in sickle cell disease: indications, advantages and potential complications. Forum Med Suisse 2006;6 (suppl 30): p64S, P142, 2006.
22. Pileggi C, Di Sanzo M, Mascaro V, Marafioti MG, Costanzo FS, Pavia M. Role of serum ferritin level on overall survival in patients with myelodysplastic syndromes: results of a meta-analysis of observational studies. PLoS One 2017 Jun;12(6):e0179016.
23. Taher AT, Origa R, Perrotta S, Kourakli A, Ruffo GB, Kattamis A, et al. New film-coated tablet formulation of deferasirox is well tolerated in patients with thalassemia or lower-risk MDS: results of the randomized, phase II ECLIPSE study. Am J Hematol. 2017 May;92(5):420-8.
24. Porter JB, Elalfy MS, Taher AT, Aydinok Y, Chan LL, Lee SH, et al. Efficacy and safety of deferasirox at low and high iron burdens: results from the EPIC magnetic resonance imaging substudy. Ann Hematol. 2013 Jan;92(2):211-9.
25. Pinto V, Balocco M, Ambaglio I, Derchi G, Malcovati L, Forni GL. Iron overload-related heart failure in a patient with transfusion-dependent myelodysplastic syndrome reversed by intensive combined chelation therapy. Clin Case Rep. 2015 Nov;3(11):952-4.
26. Mavrogeni S. Comparison of myocardial and hepatic iron loading, assessed by MRI T2*, in patients with myelodysplastic syndromes, thalassaemia major and controls. Blood Transfus. 2012 Apr;10(2):237-40
27. Porter JB, El-Alfy M, Viprakasit V, Giraudier S, Chan LL, Lai Y, et al. Utility of labile plasma iron and transferrin saturation in addition to serum ferritin as iron overload markers in different underlying anemias before and after deferasirox treatment. Eur J Haematol. 2016 Jan;96(1):19-26.
28. Brittenham GM. Iron-Chelating therapy for transfusional Iron Overload. N Engl J Med 2011 Jan;364(2):146-56.
29. de Swart L, Hendriks JC, van der Vorm LN, Cabantchik ZI, Evans PJ, Hod EA, et al Second international round robin for the quantification of serum non-transferrin-bound iron and labile plasma iron in patients with iron-overload disorders.Haematologica. 2016 Jan;101(1):38-45.
30. Komrokji R, Garcia-Manero G, Ades L, Prebet T, Steensma DP, Jurcic JG, et al. Sotatercept with long-term extension for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes: a phase 2, dose-ranging trial. Lancet Haematol. 2018 Feb;5(2):e63-2.
31. Platzbecker U, Germing U, Götze KS, Kiewe P, Mayer K, Chromik J, et al. Luspatercept for the treatment of anaemia in patients with lower-risk myelodysplastic syndromes (PACE-MDS): a multicentre, open-label phase 2 dose-finding study with long-term extension study. Lancet Oncol. 2017 Oct;18(10):1338-47.
32. Santini V. Of blood and bone: the sotatercept adventure. Lancet Haematol. 2018;5(2)e54-5.
33. Zhou L, Nguyen AN, Sohal D, Ying Ma J, Pahanish P, Gundabolu K, et al. Inhibition of the TGF-beta receptor I kinase promotes hematopoiesis in MDS. Blood. 2008 Oct;112(8):3434-43.
34. Casu C, Nemeth E, Rivella S. Hepcidin agonists as therapeutic tools. Blood. 2018 Apr;131(16):1790-1794.
35. Motta I, Scaramellini N, Cappellini MD. Investigational drugs in phase I and phase II clinical trials for thalassemia. Expert Opin Investig Drugs. 2017 Jul;26(7):793-802.
Received 2 Sep 2018
Accepted 1 Oct 2018